靶向BnSVP的CRISPR/Cas9基因组编辑载体的构建
2018-07-06廖芳丽赵福永
赵 恒,张 宏,廖芳丽,李 刚,赵福永
(1.长江大学 生命科学学院,湖北 荆州 434025;2.荆州市种子管理局,湖北 荆州 434020)
成簇规律间隔短回文重复序列及其相关系统CRISPR/Cas9(Clustered regularly interspaced short palindromic repeats/CRISPR-associated 9,CRISPR/Cas9 system)是继锌指核酸酶(Zinc finger nuclease,ZFN)和类转录激活因子效应物核酸酶(Transcription activator-like effector nuclease,TALEN)系统后的一种新的基因组定点编辑技术,可实现基因敲除、基因插入与定点替换、染色体重组和多基因同步敲除等功能,且具有载体构建简单,靶向特异性高等特点,是反向遗传学中研究基因功能的一种重要工具[1]。目前,CRISPR/Cas9系统已在拟南芥[2-3]、烟草[4]、杨树[5]等多种模式植物及水稻[6-7]、小麦[8-9]、玉米[10]等农作物中成功应用,在油菜中也已有报道。Yang等[11]应用CRISPR/Cas9系统以甘蓝型油菜的4个基因家族(BnaRGA、BnaFUL、BnaDA1和BnaDA2)的12个基因成员作为靶标进行了研究。结果表明,在T0,靶向单个基因的sgRNA引发的平均突变率为65.3%,而靶向多个同源基因的sgRNA组合其引发的突变率介于27.6%~96.6%,且纯合突变易于发现,没有发现脱靶现象;在T1,突变基因按孟德尔遗传规律遗传,且不存在新突变和回复突变现象。Braatz等[12]以影响甘蓝型油菜果瓣边缘发育的2个ALCATRAZ(ALC)同源基因为靶标,应用CRISPR/Cas9系统在T1筛选到1个alc双等位纯合突变株系,该株系能稳定遗传,在T2植株中未检测到野生型ALC基因;全基因组重测序发现,转化载体序列在其基因组上存在至少5次独立的插入事件,但没有检测到脱靶效应。该突变株系是一个全新的甘蓝型油菜抗裂荚种质资源。
Shortvegetativephase(SVP)是拟南芥开花时间调控网络中的一个重要转录调控因子,该基因的高表达会显著延长植株的营养生长期[13]。甘蓝型油菜SVP同源基因(BnSVP)具有9个外显子,cDNA编码区全长726 bp,翻译产物具有典型的M、I、K和C结构域,属Ⅱ型MADS-box基因[14]。应用TNDH群体,油菜开花时间调控机理研究团队在甘蓝型油菜中鉴定了4个BnSVP同源基因,分别位于A4、A9、C4和C8连锁群。为全面探讨BnSVP的生物学功能,本研究采用CRISPR/Cas9系统构建其基因组编辑载体拟对其进行敲除突变,以观测转基因株系在开花时间等表型上的变异。
1 材料和方法
1.1 质粒、菌种与试剂
植物基因编辑载体pYLCRISPR-Cas9P35S-H(GenBank登录号:KR029111)及CRISPR/sgRNA载体pYLsgRNA-AtU3b-LacZ(GenBank登录号:KR029098)和pYLsgRNA-AtU3d(GenBank登录号:KR029099)质粒DNA由湖南农业大学刘硕谦博士提供。大肠杆菌菌株TOP 10F′(LacIq)和F-DH5α感受态细胞购自上海维地生物技术公司;KOD plus酶购自东洋纺(上海)生物科技有限公司;BsaⅠ-HF酶、T4DNA 连接酶和ATP购自New England Biolabs有限公司;限制性内切酶SpeⅠ和MluⅠ购自TaKaRa生物公司;DNA纯化回收试剂盒及质粒提取试剂盒、高纯度dNTPs购自北京全式金生物技术公司。其他试剂均为分子试剂或分析纯级别。
1.2 引物设计与合成
载体构建所需引物的设计参考唐雨薇等[15]进行。靶向BnSVP特异位点的sgRNA种子序列的设计应用CRISPR-P 2.0(http://crispr.hzau.edu.cn/CRISPR2/)完成。设计流程为:首先选取U3作为sgRNA转录启动子,sgRNA种子序列默认长度为20 bp,靶标基因组为Brassicanapus(4.1),输入BnSVP基因标签号BnaC08g34920D,点击提交;然后再根据网站输出结果及sgRNA设计的一般要求筛选特异性高、脱靶效应低的DNA序列作为sgRNA种子序列。本试验所用引物序列(表1)均由上海生工生物技术公司合成。
1.3 载体质粒的转化、提取与菌种保存
取pYLCRISPR-Cas9P35S-H、pYLsgRNA-AtU3b-LacZ和pYLsgRNA-AtU3d质粒DNA原液各1 μL,并用ddH2O稀释10倍。采用热激法分别转化大肠杆菌菌株TOP 10F′(LacIq)和F-DH5α感受态细胞,分别涂布在含卡那霉素(50 mg/L)和氨苄青霉素(100 mg/L)的LB平板上,37 ℃倒置培养过夜。挑取单菌落摇菌,用质粒提取试剂盒分别提取各载体质粒DNA,用含20%灭菌甘油LB液体培养基保存各菌种。质粒DNA于-20 ℃保存备用。
1.4 sgRNA表达盒的构建
为提高打靶效率,每个靶向BnSVP的基因组编辑载体均同时包含2个靶点Targert 1(T1)和Target 2(T2)。sgRNA种子序列均选自BnSVP的外显子区(或包含部分内含子序列),位于其编码蛋白的重要功能域。
各靶点sgRNA表达盒的构建采用巢式(Nested)PCR和重叠(Overlapping)PCR技术进行扩增,需要经过2轮PCR反应,第1轮2个PCR反应,第2轮1个PCR反应。T1sgRNA的构建:第1轮反应均以pYLsgRNA-AtU3b-LacZ(2~5 ng)为扩增模板,第1个反应用U-F/SVPAU3bT1引物组合,第2 个反应用SVPAgRT1/gR-R引物组合,2个反应使用相同的15 μL PCR扩增体系[15]。扩增程序为:94 ℃,2 min;94 ℃,10 s,58 ℃,20 s,68 ℃,30 s,26个循环。扩增完毕各取3 μL反应液进行电泳检测,判断扩增片段的大小是否符合预期并估测其浓度。从上述2个反应管中各取1 μL用ddH2O稀释10倍,然后各取1 μL作为第2轮PCR扩增的模板。第2轮PCR扩增的引物组合为Pps-R-C/Pgs-GG2,扩增体系为30 μL[15]。扩增程序为:94 ℃,2 min;94 ℃,10 s,58 ℃,20 s,68 ℃,30 s,28个循环。扩增完毕,反应液用2.0%琼脂糖进行凝胶电泳,切取含预期大小目标片段凝胶用DNA纯化回收试剂盒回收。T2sgRNA的构建流程与T1sgRNA的构建流程类似,但其扩增质粒模板为pYLsgRNA-AtU3d,第1轮2个反应的引物组合分别为U-F/SVPAU3dT2和SVPAgRT2/gR-R;第2轮PCR反应的引物组合为Pps-GG2/Pgs-L-C。

表1 靶向BnSVP基因组编辑载体构建PCR引物Tab.1 PCR primers for genome editing constructs targeting BnSVP
注:序列中斜体小写字母为酶切识别序列;大写加粗字母为接头序列。
Note:Sequence in italic lowercase letters represents restriction enzyme recognition site;Sequence in bold capital letters is linker.
1.5 双靶点sgRNA表达盒与pYLCRISPR/Cas9 载体的组装
取T1sgRNA和T2sgRNA第2轮PCR回收产物各20 μL,pYLCRISPR-Cas9P35S-H质粒溶液10 μL,BsaⅠ-HF 1.5 μL(30 U),分别构建50 μL酶切体系,37 ℃酶切30 min。酶切产物用2.0%琼脂糖凝胶进行电泳,切取目的条带用凝胶回收试剂盒进行回收,各取3 μL回收液进行电泳并估测浓度。然后按照载体:靶标分子数为1∶3的比例构建10 μL连接反应体系,16 ℃连接过夜。
1.6 重组载体的转化与检测
从-80 ℃冰箱中取1管(100 μL)F-DH5α感受态细胞,冰上化冻,取10 μL上述连接产物加入其中,轻弹混匀,冰浴静置20 min;42 ℃水浴热激45 s,迅速将管子置冰中2 min,加入平衡至室温不含抗生素的LB液体培养基700 μL;37 ℃,200 r/min,振荡培养30 min;5 000 r/min离心1 min,留取100 μL上清并重悬浮菌块,然后涂布于含25 mg/L Kan、200 mg/L IPTG和20 mg/mL X-gal 的LB平板,37 ℃倒置培养过夜。挑取蓝斑菌落利用测序引物组合(SP-L2/SP-R-C)进行菌落PCR。将阳性菌落接种于5 mL 含50 mg/L Kan 的LB 液体培养基中,37 ℃,150 r/min,振荡培养过夜,用质粒提取试剂盒分别提取质粒,SpeⅠ和MluⅠ双酶切进行进一步验证,挑取正确质粒送上海生工生物技术公司进行测序。
2 结果与分析
2.1 靶向BnSVP的sgRNA设计
靶向特定基因或DNA片段的sgRNA由两部分组成,即5′端的种子序列(约20 bp)和3′端保守的骨架序列(83 bp),因此,载体构建时仅需对20 bp的种子序列进行设计。用宁油7号SVP基因序列对甘蓝型油菜参考基因组序列(v4.1)进行同源搜索,共获得4个BnSVP同源基因(BnaA09g42480D、BnaC08g34920D、BnaC04g35060D、LOC106446430),与本团队前期利用TNDH群体对该基因的遗传定位结果一致。应用Lasergene 7.1对序列进行分析发现,BnSVP.A09(BnaA09g42480D)与BnSVP.C08(BnaC08g34920D)cDNA的序列相似性为97.5%,氨基酸一致性为98.3%;BnSVP.A04(LOC106446430)与BnSVP.C04(BnaC04g35060D)cDNA的序列相似性为98.9%,氨基酸一致性为98.8%。而前二者和后二者之间的cDNA的序列相似性在90.8%~92.3%,氨基酸一致性在91.7%~92.5%,因此,4个BnSVP可以归为2个亚组,推测各个亚组内的基因成员在功能上更接近。因此,针对各亚组基因成员及全部4个BnSVP基因,应用CRISPR-P 2.0(http://crispr.hzau.edu.cn/CRISPR2/)在线设计了12条sgRNA种子序列(表1、图1)。

序列为BnSVP的cDNA编码区及部分内含子,大写字母代表外显子,符号*表示其边界;小写字母为内含子序列;GG(+)/CC(-)表示PAM识别位点,下划线标记序列为靶向BnSVP的sgRNA的种子序列。Each sequence includes coding region and partial intron of BnSVP,Capital letters represent exonsand star symbols definite their borders, while lowercase letters represents intron sequences;GG(+)/CC(-)means the recognition PAM site of the Cas9,the sequence with underline means the candidate targeting sequence of BnSVP.
2.2 靶向BnSVP的sgRNA表达盒的扩增
取pYLsgRNA-AtU3b-LacZ和pYLsgRNA-AtU3d质粒DNA各1 μL,并用ddH2O稀释100倍,分别用作T1sgRNA和T2sgRNA表达盒构建PCR扩增模板。第1轮PCR引物组合U-F/SVPAU3bT1扩增含LacZ-AtU3b片段,大小为580 bp,引物组合SVPAgRT1/gR-R扩增T1sgRNA片段,大小为136 bp;引物组合U-F/SVPAU3dT2扩增含AtU3d片段,大小为155 bp,引物组合SVPAgRT2/gR-R扩增T2sgRNA片段,大小为135 bp(图2-A)。第2轮PCR采用重叠PCR策略组装各靶标sgRNA,T1sgRNA扩增产物长713 bp,T2sgRNA扩增产物长289 bp(图2-B)。
2.3 sgRNA表达盒与CRISPR/Cas9载体的组装
sgRNA表达盒与CRISPR/Cas9载体的组装采用Golden gate cloning策略[16]。在各靶标sgRNA构建的第2轮PCR所使用的引物中已设计有BsaⅠ-HF酶切识别序列(GGTCTC)和连接头序列(表1),CRISPR/Cas9载体和T1sgRNA及T2sgRNA DNA经BsaⅠ-HF酶切后将会在各片段之间产生首尾兼容的4碱基5′突出末端(图3)。sgRNA表达盒酶切片段与CRISPR/Cas9载体按正确顺序连接的质粒,其转化菌落在含Kan、IPTG和X-gal 的LB平板上会显蓝色。首先,挑取蓝色单菌落用测序引物(SP-L2/SP-R-C)进行菌落PCR,并同时进行接种摇菌。正确组装的质粒载体其扩增产物大小为1 102 bp,而原质粒载体的扩增产物为825 bp(图4-A)。选取菌落PCR扩增产物大小符合预期的菌液提取质粒DNA,进一步用SpeⅠ和MluⅠ进行双酶切检测,正确组装的基因组编辑载体能切出一条936 bp的片段,而原质粒载体上无此两限制性内切酶识别位点(图4-B)。综合Kan筛选、蓝白斑筛选、菌落PCR和双酶切检测结果,本研究所构建的靶向BnSVP的6个基因组编辑载体均获得了正确连接的质粒DNA,测序结果如图5所示。

A. 第1轮PCR扩增;B. 第2轮PCR扩增;M. 100 bp ladder DNA标准分子量;-. 阴性对照;1. AtU3d;2. T2sgRNA;3. LacZ-AtU3b;4. T1sgRNA;5. T2sgRNA 表达盒;6. T1sgRNA 表达盒。
A. 1st round PCR;B. 2nd round PCR;M. 100 bp ladder DNA Marker;-. Negative control;1. AtU3d;2. T2sgRNA;3. LacZ-AtU3b;4. T1sgRNA;5. T2sgRNA expression cassette;6. T1sgRNA expression cassette.
图2sgRNA表达盒构建PCR扩增产物电泳结果
Fig.2PCRfragmentsofsgRNAexpressioncassetteconstruction
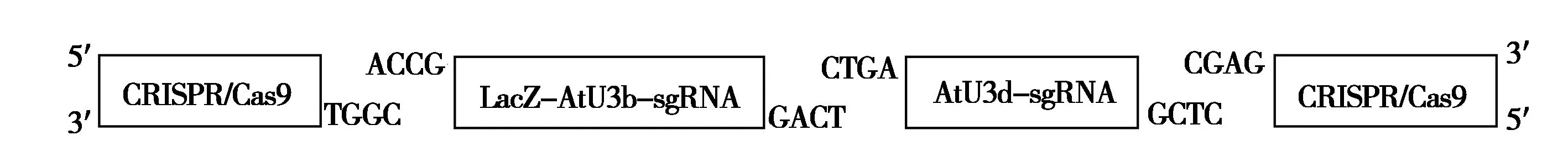
图3 sgRNA表达盒与CRISPR/Cas9载体组装示意图Fig.3 Diagram of dual sgRNA expression cassettes assembling with CRISPR/Cas9 vector

A.菌落PCR;B.双酶切;M.100 bp ladder DNA标准分子量;1.pYLCRISPR-Cas9P35S-H质粒;2~6.蓝斑菌落;7.pYLCRISPR-Cas9P35S-H-SVPA-T1/T2质粒;8. pYLCRISPR-Cas9P35S-H-SVPA-T3/T4质粒;9.pYLCRISPR-Cas9P35S-H质粒。
A. Colony PCR;B. Double digestion;M. 100 bp ladder DNA Marker;1. pYLCRISPR-Cas9P35S-H plasmid;2-6. Blue colonies;7. pYLCRISPR-Cas9P35S-H-SVPA-T1/T2 DNA;8. pYLCRISPR-Cas9P35S-H-SVPA-T3/T4 DNA;9. pYLCRISPR-Cas9P35S-H DNA.
图4CRISPR/Cas9重组载体菌落PCR及双酶切检测
Fig.4ColonyPCRanddoubledigestiondetectionsofrecombinantCRISPR/Cas9vectors
3 讨论
3.1 靶向多拷贝同源基因sgRNA的设计
CRISPR/Cas9系统是一种高效的植物基因组编辑工具。Cas9蛋白在sgRNA引导下能在特定位点切割DNA形成双链断口(Double-strand breaks,DSBs),从而激活细胞自身修复机制,并通过同源重组(Homologous recombination,HR)或非同源末端连接(Non-homologous end joining,NHEJ)造成特定位点碱基的插入或缺失,实现基因敲除。该系统的特异性主要决定于靶sgRNA中5′端约20 bp长的种子序列[17]。对于具有参考基因组序列的物种,sgRNA种子序列的设计相对简单,尤其是靶向单拷贝基因的,但对多基因家族或多拷贝同源基因的特异sgRNA的设计则相对困难。

图5 同时靶向4个BnSVP的CRISPR/Cas9重组载体测序结果Fig.5 Sequencing results of recombinant CRISPR/Cas9-based vectors targeting four BnSVP homologues simultaneously
甘蓝型油菜是异源四倍体(AACC,2n=38),其基因组中多数基因同源拷贝数为4~6个,且其DNA序列相似度极高[18]。Yang等[11]研究表明,由于序列相似度达98.22%,无法设计特异的sgRNA用以区分BnaDA2家族的2个成员(BnaA2.DA2.1、BnaA2.DA2.2)。本研究在设计靶向4个BnSVP同源基因的特异sgRNA时,充分利用了其DNA正负链上的PAM位点,并应用了双靶点组合策略,能够实现4个或2个同源基因同时敲除,但其编辑效率还有待用原生质体瞬时表达系统进行进一步检验。
3.2 TsgRNA与CRISPR/Cas9载体的组装
pYLCRISPR-Cas9P35S-H载体含有1个ccdB致死基因,而在ccdB表达盒两侧各有1个BsaⅠ酶切位点。经BsaⅠ处理后,ccdB表达盒(685 bp)被切除,线性化载体的两端分别形成了具有CGGT和CGAG的4碱基5′突出黏性末端[18]。T1sgRNA和T2sgRNA两末端均设计有BsaⅠ酶切位点,经BsaⅠ处理后,可形成与载体末端互补的4碱基黏性末端,在DNA连接酶的作用下可重新环化。本研究分别采用2种方法对双靶点TsgRNA与CRISPR/Cas9载体的组装效率进行了比较分析,大肠杆菌转化试验结果表明,酶切连接同体系的一步法比先酶切再连接的二步法的组装效率略低,但2种方法均能获得正确组装的阳性克隆子。菌落PCR结果证明,抗性蓝斑菌落并非全部是正确组装的,而抗性白斑菌落也并非都是非正确组装的,且白斑菌落中仍然存在含ccdB基因的非重组型质粒,理论上含ccdB基因的质粒和非LacIq基因型的宿主细胞是不兼容的。其主要原因可能与BsaⅠ的酶切错误及之后的错误连接有关[16]。此外,转化产物进行Kan筛选时,平板中Kan浓度不宜过高,本研究发现,以25 mg/L为最佳。若早期使用较高的Kan浓度则较难获得转化菌落,这可能与pYLCRISPR-Cas9P35S-H的低拷贝特性有关。
CRISPR-Cas9系统具有简便高效的特点,是当前植物基因组编辑技术的首选,已在植物功能基因组学研究和分子育种研究领域展现了广阔的应用前景。随着该系统的不断完善和创新[19-20],必将引领新一轮的植物分子生物学技术革命。
参考文献:
[1] 单奇伟,高彩霞. 植物基因组编辑及衍生技术最新研究进展[J]. 遗传,2015,37(10):953-973.
[2] Schiml S,Fauser F,Puchta H. The CRISPR/Cas system can be used as nuclease for in planta gene targeting and as paired nickases for directed mutagenesisinArabidopsisresulting in heritable progeny[J]. The Plant Journal :for Cell and Molecular Biology,2014,80(6):1139-1150.
[3] Hilton I B,D′ippolito A M,Vockley C M,et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers[J]. Nature Biotechnology,2015,33(5):510-517.
[4] Gao J,Wang G,Ma S,et al. CRISPR/Cas9-mediated targeted mutagenesis inNicotianatabacum[J]. Plant Molecular Biology,2015,87(1/2):99-110.
[5] Fan D,Liu T,Li C,et al. Efficient CRISPR/Cas9-mediated targeted mutagenesis in populus in the first generation[J]. Scientific Reports,2015,5:12217.
[6] Zhou H,Liu B,Weeks D P,et al. Large chromosomal deletions and heritable small genetic changes induced by CRISPR/Cas9 in rice[J]. Nucleic Acids Research,2014,42(17):10903-10914.
[7] Endo M,Mikami M,Toki S. Multigene knockout utilizing off-target mutations of the CRISPR/Cas9 system in rice[J]. Plant & Cell Physiology,2015,56(1):41-47.
[8] Upadhyay S K,Kumar J,Alok A,et al. RNA-Guided genome editing for target gene mutations in wheat[J]. G3-Genes Genomes Genetics,2013,3(12):2233-2238.
[9] Zhang Y,Liang Z,Zong Y,et al. Efficient and transgene-free genome editing in wheat through transient expression of CRISPR/Cas9 DNA or RNA [J]. Nature Communications,2016,7:12617.
[10] Svitashev S,Young J K,Schwartz C,et al. Targeted mutagenesis,precise gene editing,and Site-Specific gene insertion in maize using Cas9 and guide RNA[J]. Plant Physiology,2015,169(2):931-945.
[11] Yang H,Wu J J,Tang T,et al. CRISPR/Cas9-mediated genome editing efficiently creates specific mutations at multiple loci using one sgRNA inBrassicanapus[J]. Scientific Reports,2017,7(1):7489.
[12] Braatz J,Harloff H J,Mascher M,et al. CRISPR-Cas9 targeted mutagenesis leads to simultaneous modification of different homoeologous gene copies in polyploid oilseed rape(Brassicanapus)[J]. Plant Physiology,2017,174(2):935-942.
[13] Hartmann U,Höhmann S,Nettesheim K,et al. Molecular cloning of SVP:a negative regulator of the floral transition inArabidopsis[J]. The Plant Journal:for Cell and Molecular Biology,2000,21(4):351-360.
[14] 潘丹丹,毛群飞,张金顺,等. 油菜开花时间调控基因SVP的克隆与表达特性分析[J]. 华北农学报,2013,28(1):93-100.
[15] 唐雨薇,刘丽萍,王若娴,等. 茶树咖啡碱合成酶CRISPR/Cas9基因组编辑载体的构建[J]. 茶叶科学,2016,36(4):414-426.
[16] Taylor G P. Current Protocols in Molecular Biology [M]//Ma X,Liu Y G. CRISPR/Cas9-based multiplex genome editing in monocot and dicot plants. New Jersey:John Wiley & Sons,Inc.,2016,115:31.6.1-31.6.21.
[17] Ding Y,Li H,Chen L L,et al. Recent advances in genome editing using CRISPR/Cas9[J].Frontiers in Plant Science,2016,7:703.
[18] Lysak M A,Koch M A,Pecinka A,et al. Chromosome triplication found across the tribeBrassiceae[J]. Genome Research,2005,15(4):516-525.
[19] Lu H P,Liu S M,Xu S L,et al. CRISPR-S:an active interference element for a rapid and inexpensive selection of genome-edited,transgene-free rice plants[J]. Plant Biotechnology Journal,2017,15(11):1371-1373.
[20] Ren B,Yan F,Kuang Y,et al. Improved base editor for efficiently inducing genetic variations in rice with CRISPR/Cas9-guided hyperactive hAID mutant[J]. Molecular Plant,2018,11(4):623-626.
