Wharton’s jelly derived allogeneic mesenchymal stromal cells for treatment of type 1 diabetes: study protocol for a double-blinded, randomized, parallel,placebo-controlled trial
2018-07-04
1 Department of Medical Cell Biology, Uppsala University, Uppsala, Sweden
2 Department of Medical Sciences, Uppsala University, Uppsala, Sweden
3 Karolinska Trial Alliance, Karolinska University Hospital, Huddinge, Sweden
4 NextCell Pharma AB, Huddinge, Sweden
5 Department of Laboratory Medicine, Karolinska Institutet, Huddinge, Sweden
INTRODUCTION
Type 1 diabetes (T1D) is an insulin dependent, autoimmune disorder resulted from T cell mediated destruction of insulinproducing pancreatic beta-cells. At onset of T1D, beta-cell mass has regularly decreased to 20–40% compared to normal levels. Maintenance of residual insulin secretion is important for contributing to markedly lower hemoglobin A1c (HbA1c), less blood glucose fluctuations, and diminished risk of ketoacidosis. It also substantially decreases the risks of developing severe hypoglycemic events and late complications.1,2An identified successful intervention may not only be used to prevent disease but also be applied to patients with ongoing disease before overt hyperglycemia, thereby providing means to halt disease progression.
Different intervention strategies have been tested to save residual beta-cells, but at best only endogenous insulin production has been temporarily preserved.
Wharton’s jelly derived mesenchymal stromal cells(WJMSCs) are known to have immunoprivileged characteristics. Thus, they express low levels of major histocompatibility complex (MHC) class I antigens and seem to be essentially devoid of class II antigens. Similar to trophoblasts, they have been shown to express human leukocyte antigen G (HLA-G), which may influence immunological properties. They also synthesize immunosuppressive molecules such as indoleamine-2,3-dioxygenase (IDO)and prostaglandin E2.3The immunosuppressive functions of mesenchymal stromal cells (MSCs) are triggered by the surrounding microenvironment, where abundant inflammatory factors are released by immune cells.4Detailed investigations addressing how the microenvironment affects homing and immunosuppressive effects of MSCs are still lacking.
The purposes of this study are to determine whether intravenous infusion of allogeneic WJMSCs is safe in adult patients with T1D and to investigate changes in beta-cell function, metabolic control, and diabetes treatment satisfaction during one year study period. The study population will consist of adult patients with T1D (< 2 years), 18–40 years of age(inclusive at both ends), only male in the first stage and both sexes in the second stage of the trial.
No major adverse events have been linked to administration of allogeneic MSCs, irrespective of cell source and treatment indication, but an anti-donor response has been observed. Both the POSEIDON trial (A Phase I/II, Randomized Pilot Study of the Comparative Safety and Efficacy of Transendocardial Injection of Autologous Mesenchymal Stem Cells Versus Allogeneic Mesenchymal Stem Cells in Patients With Chronic Ischemic Left Ventricular Dysfunction Secondary to Myocardial Infarction) and a recent phase 2 Mesoblast trial reported generation of anti-donor antibodies in 13% of patients treated with allogeneic MSC from bone marrow. The first stage of the trial will only accept male study subjects to eliminate the risk of human leukocyte antigen immunization of fertile women. After this risk has been evaluated, both genders will be included in the second stage.5,6
Since the study aims to preserve remaining insulin production, all patients eligible for study must have a fasting C-peptide concentration > 0.12 nM. Patients aged > 40 years often have a different progression of disease from that in younger individuals, with very slow loss of insulin production and no insulin-dependence at diagnosis (sometimes denoted as Latent Autoimmune Diabetes in the Adult (LADA)). Many of these patients show strong similarities to type 2 diabetes patients.7
To investigate the effects of intervention on T1D development in a study population without a strongly heterogeneous issue,we only include patients aged 18–40 years. In line with theDeclaration of Helsinki, it is reasonable to first provide a safety and proof of principle study in young adults. Studies in children and adolescents would also require a stratified analysis with regards to beta-cell function evaluations, since progression of disease in children and adolescents is even more aggressive than in young adults.8
METHODS/DESIGN
Study design
This is a two-stage design study. An open, non-randomized,dose-escalation scheme will be used in the first stage of the study, and a randomized, double-blinded, parallel, placebocontrolled scheme in the second stage of the study (Figure 1).
Eligibility criteria
Inclusion criteria
Patients with T1D meeting all of the following conditions are considered for inclusion:
Strengths and limitations
Strengths
- There are few prospective, randomized, doubleblinded, place-controlled trials with mesenchymal stromal cells and in particular with Wharton’s jelly derived allogeneic mesenchymal stromal cells. Our trial has a robust design and will provide the highest level of evidence.
- The manufacturing process of the investigational medicinal product has a high batch yield with low batch to batch variation. This is due to the selection algorithm and the pooling of multiple donors. A robust manufacture with low variation between batches facilities the possibility to detect efficacy.
Limitations
- The age of patients will not reflect the typical age group of newly diagnosed T1D patients. However, in the first stage of the study for safety data, we will only include patients aged 18 years or older.
- Three different doses will be evaluated for safety in the first stage of the trial. Only three patients in each dose cohort will not generate any statistically significant efficacy data.
- Efficacy will be evaluated in the second stage but the patient number is low and the power calculations are based on a trial using autologous bone marrow derived MSCs.
1. Provision of written informed consent before undergoing any study-specific procedures
2. Clinical history compatible with T1D (clinical diagnosis)diagnosed less than 2 years before enrolment
3. In the first stage of the study, only male patients (patients 1–9) at 18–40 years old will be included. In the second stage of the study, both male and female patients (patients 10–24) at 18–40 years old will be included
4. Mentally stable and, in the opinion of the investigator,ability to comply with the procedures of the study protocol
5. Fasting plasma C-peptide concentration > 0.12 nM
6. Women of child-bearing potential, defined as all women physiologically capable of becoming pregnant, if they are using effective methods of contraception during the study period. Acceptable birth control methods are those with a failure rate of < 1% per year when used consistently and correctly. (Specified in “Recommendations related to contraception and pregnancy testing in clinical trials”,supplied from www.hma.eu/)
Exclusion criteria
Patients with T1D meeting one or more of the following conditions are excluded from this study:
1. Inability to provide informed consent
2. Patients with body mass index (BMI) > 30 kg/m2, or weight> 100 kg
3. Patients with weight < 50 kg

Figure 1: Study flowchart.
4. Patients with unstable cardiovascular status including New York Heart Association (NYHA) Functional Classification class III/IV or symptoms of angina pectoris
5. Patients with uncontrolled hypertension (≥ 160/105 mmHg)
6. Patients with active infections unless treatment is not judged necessary by the investigators
7. Patients undergoing latent or previous as well as on-going therapy against tuberculosis, or exposed to tuberculosis or have travelled in areas with high risk of tuberculosis or mycosis within the last 3 months
8. Patients with serological evidence of infection with human immunodeficiency virus, Treponema pallidum, hepatitis B antigen (patients with serology consistent with previous vaccination and a history of vaccination are acceptable) or hepatitis C
9. Patients with any immunosuppressive therapy
10. Patients with known demyelinating disease or with symptoms or physical examination findings consistent with possible demyelinating disease
11. Pregnant or nursing (lactating) women, where pregnancy is defined as the state of a female after conception and until the termination of gestation, confirmed by a positive human chorionic gonadotrophin (hCG) laboratory test
12. Patients with known, or previous, malignancy
13. Taking oral anti-diabetic therapies or any other concomitant medication which may interfere with glucose regulation other than insulin
14. Patients with glomerular filtration rate (GFR) <80 mL/min/1.73 m2body surface
15. Patients with proliferative retinopathy
16. Patients with any condition or any circumstance that in the opinion of the investigator would make patients unsafe to undergo treatment with MSCs
17. Known hypersensitivity reaction to any excipients,i.e.,dimethyl sulfoxide (DMSO)
Withdrawal and elimination criteria
Patients are free to discontinue their participation in the study at any time for any reason without affecting their right to appropriate follow-up investigation and future treatment.
Patients may be discontinued from the study at any time at the discretion of the investigator. Specific reasons for discontinuing a patient from further assessments are specified in the protocol.
Randomization and blinding
Randomization and blinding are only applicable for the second stage of the trial and will be performed with a web based randomization system. The randomization code will be created using a computer-generated randomization schedule prepared prior to the start of the study. The randomization will be made in blocks of three (batch 1, batch 2, and placebo). The different batches and placebo will have codes and the key is kept secret for both investigators and patients. Unblinding is conducted after end of trial.
Intervention
Investigational medicinal product (IMP)
The manufacturer Polski Bank Komorek Macierzystych S.A. (PBKM) (Poland) receives donated umbilical cords from Szpital Położniczo (Poland). PBKM holds a production permit issued from Polish Medicines Inspectorate for production of ATMP for clinical trials with authorization number 239/0436/17. Cells from each donor will be analyzed and expanded separately. After being washed from substrate residuals and metabolic products, quality control analyses are performed to measure concentration of cells, sterility, viability,endotoxin, mycoplasma and phenotypic expression. PBKM sends cell samples to NextCell Pharma AB (the Sponsor)who will notify which of the cells in manufacturing should be used for the formulation of the IMP (ProTrans) based on the company’s proprietary selection algorithm. The product is prepared in advance before any subject is included in the trial and provided as cryopreserved.
The cells are frozen in cryo bags at concentrations of 2 × 107cells in 5% human serum albumin and 10% DMSO, and one cryo bag contains one dose. The bags are frozen in a controlled rate freezer and directly transferred to -190ºC for storage until it is time for infusion. Further details of the manufacturing process are described in the Investigator’s Brochure and Investigational Medicinal Product Dossier.
Selection algorithm
The sponsor (NextCell Pharma AB) has developed a selection algorithm based on functional assays. The algorithm determines which cells and donors should be used for formulation of ProTrans (the IMP of the trial). The construction of this proprietary selection algorithm and which analyses are conducted is a trade secret. However, it is important to indicate that only cells and donors that pass the quality criteria of the manufacturer are considered in the selection and that all basic criteria for MSCs for clinical trial purposes are fulfilled.
Administration
IMP or placebo will be given as an intravenous infusion using a peripheral venous catheter placed in a vein on the hand or arm.
Dosage
In the first stage of the trial, patients 1–3 will receive a single dose of 25 million cells, patients 4–6 will receive 100 million cells and patients 7–9 will receive 200 million cells. In the second stage of the study, all patients will receive a fixed single dose, which is preliminarily planned to 100 million WJMSCs in saline with 5% human serum albumin (HSA) and 10%DMSO or placebo consisting of saline with 5% HSA and 10%DMSO undiluted. Prior to infusion, all cells are diluted in 100 mL saline (NaCl, Baxter) which gives a final concentration of 0.95 × 106cells/mL, 9 mg/mL sodium chloride, 0.2% human albumin and 0.5% DMSO.
Outcome measures
Primary objective
The safety and tolerance of intravenous infusion of allogeneic WJMSCs in adult patients with T1D.
Secondary objectives
Changes in beta-cell function, metabolic control, and diabetes treatment satisfaction during one year study period.
Primary safety endpoint
Safety parameters evaluated are adverse events, such as hypoglycemic episodes and allergic reactions. Besides anamnestic reports, ophthalmologic examination, ECG, vital signs, and laboratory assessments will be performed.
Primary efficacy endpoint
Delta-change (Δ-change) of C-peptide area under the curve(AUC) (0–120 minutes) for mixed meal tolerance test(MMTT) on day 372 following WJMSC/placebo infusion when compared to test performed before start of treatment.Visit schedule is depicted inTable 1.
Data collection and management
Data management based on Good Clinical Practice (GCP)refers to the activities defined to achieve safe routines to efficiently enter subject information into a database, avoiding errors. The principal investigator is responsible for ensuring the accuracy, completeness and legibility of the data reported in the case report form (CRF). This will be inspected by the monitor on a regular basis before CRFs are submitted for data entry. The investigator will maintain source documents for each patient in the study. All information on CRFs will be traceable to these source documents in the patient’s file, with the exemption of data where CRF is used as a source. The data will be verified for missing data and inconsistencies, and for any necessary medical clarifications by the investigator.
Sample size calculation
As this is primarily a safety study, only 24 patients will be included and undergo a dose escalation scheme. The second blinded part will be composed of 10 patients treated with 2 batches of WJMSCs and 5 placebo treated patients. A calculation of power and patient numbers has been performed for the second stage of the study, based on results for similar primary outcome variable from a previous study where patients were treated with autologous bone marrow-derived MSC (Delta-change of C-peptide area under the curve (0–120 minutes) for mixed meal tolerance test on day 372 following WJMSC/Placebo infusion when compared to test performed before start of treatment) and type of patients presented inFigure 1of the article written by Carlsson et al.9Patients in the described previous study were treated with autologous bone marrow-derived MSCs, and we expect similar results, although this remains to be tested. For calculation of patient numbers,the second stage of the study was considered to contain three randomized groups (placebo, batch 1 and batch 2), and not to include any data from the first stage of the study (since this stage of the study is not randomized),n= 5 per group gives a power of 95 % and analysis of variance of the three groups should be significant and allow post hoc comparison between groups.
Statistical analysis
Efficacy endpoints will all be evaluated as Δ-changes using Student’s unpaired two-tailedt-test. The full analysis set(PPS; for definition, see below) will be evaluated for all endpoints. Treatment difference will be estimated with 95%confidence interval. Non-normally distributed data with two nominal variables in exploratory endpoints will be analyzed by Wilcoxon’s signed rank sum test. Calculations will be performed in Prism (Graphpad Software, San Diego, CA,USA).
Trial quality assurance and control
A monitor from Karolinska Trial Alliance is responsible for coordinating the activities of the study and ensures adherence to The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH)guidelines. Before study initiation, the protocol and CRFs were reviewed with investigator and staff. The monitor will visit the site during the course of study in order to verify adherence to the clinical study protocol, completeness and accuracy of data entered to the CRF by comparing them with the source documents. The monitor will review the progress of the clinical investigation, and review CRFs and original source data/documents with the clinical investigational personnel, for accuracy of data recording, and facilities used in the clinical investigation (e.g., local laboratory) may be reviewed. Visits will be documented in a monitory log in the investigator’s file and a monitor report for each visit will be sent to and signed by the sponsor.
Ethics and dissemination
This study protocol has been approved by the Ethics Committee of Stockholm (approval number 2017/1533-31/2)(Additional file 1), and will be performed in accordance with theDeclaration of Helsinki. Written informed consent will be obtained from each patient (Additional file 2).
We will publish the findings in national and internationaljournals, and present these at national and international conferences. The protocol adheres to the recommendations provided by the SPIRIT 2013.10
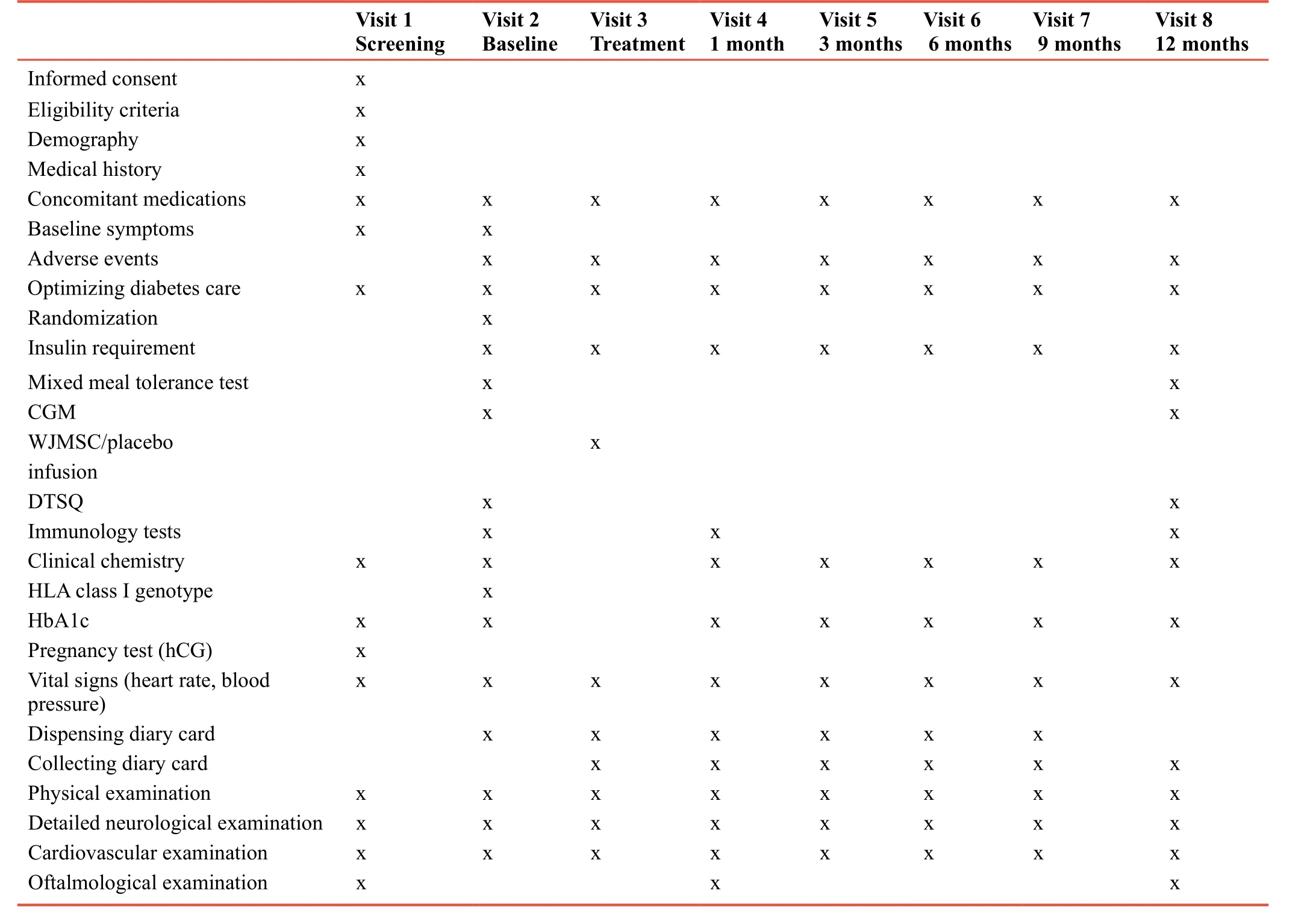
Table 1: Complete list of outcome measures and assessments
DISCUSSION
No or only minor side effects of MSCs have been observed in the clinical treatment of graft versus host disease, myocardial infarct, liver cirrhosis, or osteogenesis imperfecta. No increased risk of tumor development in patients has been reported, and no ectopic tissue formation has been observed.11,12Similarly,in the first studies in patients with T1D, no side effects were observed.9,13El-Badawy and El-Badri reported a meta-analysis of 22 studies where T1D was treated with stem cell therapy.14They concluded that various stem cell therapies seemed to be safe and could potentially lead to remission of diabetes.
There are three key components to treat T1D: to reduce hyperglycemia, to relieve autoimmunity and to regenerate lost islets. With strong evidence of immunomodulatory effect and transdifferentiation potential, MSCs can regulate the autoreactive host immune system and promote islet regeneration to replenish the lost islets. MSCs can alleviate self-reactive lymphocytes-caused inflammatory damage to newly-formed beta-cells because of their immunomodulatory properties. The ability of MSCs to secrete trophic and angiogenic factors may also restrict islet damage by establishing a microenvironment which stimulates beta-cell growth, survival, and differentiation. There is an increased need to describe the mechanisms of MSC-mediated cell therapy in detail. Well-designed large scale randomized studies considering the stem cell type, cell number, and infusion method in T1D patients are also urgently needed.
WJMSC from this manufacturer has been used in hospital exemption procedures under various conditions and the safety profile of the cells is consistent and well tolerated with only mild and transient adverse reactions related to the product.
However, serious adverse events and deaths have been reported in patients with terminal amyotrophic lateral sclerosis or grade 4 graft versus host disease who undergo WJMSCs therapy.
The rational of ProTrans is to utilize the selection algorithm,developed by NextCell Pharma AB, to identify and select suitable donors and/or optimal MSC populations for formulation of a potent stem cell product with low batch to batch variability and sustained, well tolerated safety profile.
TRIAL STATUS
Patient recruitment was initiated in January 2018 for the first part of the trial,i.e., the dose escalation. As of June 1, 2018 all three patients in the low-dose cohort and all three patients in the medium-dose cohort had been treated. The first part of the trial is expected to be completed to collect 1-month follow-up safety data in October 2018. Primary outcome measures will be estimated in 2020. No serious adverse events had been recorded on June 1, 2018. Patients treated in the low- and medium-dose cohorts had not suffered from any adverse events during the treatment. At the 1-month follow-up visit,only mild adverse events were reported, without any causality with treatment.
Additional files
Additional file 1: Ethical Approval Documentation.
Additional file 2: Model consent form.
Author contributions
POC is the principle investigator. POC and MGS are responsible for the design of the study and the study protocol. MGS drafted the protocol manuscript and POC revised it. Both authors have read and approved the final protocol manuscript.
Conflicts of interest
MGS is employed by NextCell Pharma AB and own shares in the company.
Financial support
This study is sponsored by NextCell Pharma AB.
Institutional review board statement
This study protocol was approved by the Ethics Committee Stockholm(approval number 2017/1533-31/2) and the Swedish Medicinal Product Agency (EudraCT number: 2017-002766-50), and will be performed in accordance with theDeclaration of Helsinki.
Declaration of patient consent
The authors certify that they will obtain all appropriate patient consent forms. In the form the patients will give their consent for their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Reporting statement
This study follows the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) guidance for protocol reporting.
Biostatistics statement
The statistical methods of this study were reviewed by the biostatistician of Karolinska Trial Alliance, Karolinska University Hospital in Sweden.
Copyright transfer agreement
The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement
Dissemination plans include presentations at scientific conferences and scientific publications. Individual participant data will not be available.However, the study protocol and informed consent form will be made available beginning 3 months and ending 5 years following article publication to investigators whose proposed use of the data has been approved by an independent review committee identified to achieve aims in the approved proposal. In order to gain access, data requestors will need to sign a data access agreement. Proposals should be directed to mathias.svahn@nextcellpharma.com.
Plagiarism check
Checked twice by iThenticate.
Peer review
Externally peer reviewed.
Open access statement
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
REFERENCES
1. Madsbad S, Alberti KG, Binder C, et al. Role of residual insulin secretion in protecting against ketoacidosis in insulindependent diabetes.Br Med J. 1979;2:1257-1259.
2. Steffes MW, Sibley S, Jackson M, Thomas W. Beta-cell function and the development of diabetes-related complications in the diabetes control and complications trial.Diabetes Care.2003;26:832-836.
3. Kalaszczynska I, Ferdyn K. Wharton's jelly derived mesenchymal stem cells: future of regenerative medicine?Recent findings and clinical significance.Biomed Res Int.2015;2015:430847.
4. Ren G, Zhang L, Zhao X, et al. Mesenchymal stem cell -mediated immunosuppression occurs via concerted action of chemokines and nitric oxide.Cell Stem Cell.2008;2:141-150.
5. Ankrum JA, Ong JF, Karp JM. Mesenchymal stem cells:immune evasive, not immune privileged.Nat Biotechnol.2014;32:252-260.
6. Lalu MM, McIntyre L, Pugliese C, et al. Safety of cell therapy with mesenchymal stromal cells (SafeCell): a systematic review and meta-analysis of clinical trials.PLoS One.2012;7:e47559.
7. Hjort R, Alfredsson L, Carlsson PO, et al. Low birthweight is associated with an increased risk of LADA and type 2 diabetes:results from a Swedish case-control study.Diabetologia.2015;58:2525-2532.
8. Greenbaum CJ, Beam CA, Boulware D, et al. Fall in C-peptide during first 2 years from diagnosis: evidence of at least two distinct phases from composite Type 1 Diabetes TrialNet data.Diabetes.2012;61:2066-2073.
9. Carlsson PO, Schwarcz E, Korsgren O, Le Blanc K. Preserved beta-cell function in type 1 diabetes by mesenchymal stromal cells.Diabetes.2015;64:587-592.
10. Chan AW, Tetzlaff JM, Altman DG, et al. SPIRIT 2013 Statement: defining standard protocol items for clinical trials.Ann Intern Med.2013;158:200.
11. von Bahr L, Batsis I, Moll G, et al. Analysis of tissues following mesenchymal stromal cell therapy in humans indicates limited long-term engraftment and no ectopic tissue formation.Stem Cells.2012;30:1575-1578.
12. von Bahr L, Sundberg B, Lonnies L, et al. Long-term complications, immunologic effects, and role of passage for outcome in mesenchymal stromal cell therapy.Biol Blood Marrow Transplant.2012;18:557-564.
13. Hu J, Yu X, Wang Z, et al. Long term effects of the implantation of Wharton's jelly-derived mesenchymal stem cells from the umbilical cord for newly-onset type 1 diabetes mellitus.Endocr J.2013;60:347-357.
14. El-Badawy A, El-Badri N. Clinical efficacy of stem cell therapy for diabetes mellitus: a meta-analysis.PLoS One.2016;11:e0151938.
杂志排行

Clinical Trials in Degenerative Diseases的其它文章
- Fenestration and debridement combined with percutaneous minimally invasive fibula implantation in the treatment of senile degenerative osteonecrosis of the femoral head: a study protocol for a nonrandomized, controlled, clinical trial
- Percutaneous transforaminal endoscopic discectomy for treatment of degenerative lumbar disc herniation in older adult patients: study protocol for a randomized controlled trial and preliminary results
- Rapamycin-eluting stents for unprotected left main coronary artery stenosis in older adult patients with coronary atherosclerosis: study protocol for a prospective, non-randomized, controlled trial and preliminary results
- Three methods for reducing back pain in older adults with age-related osteoporotic vertebral compression fractures of the thoracolumbar spine: protocol for a non-randomized controlled trial with 2-year follow-up and preliminary results
- Deep brain stimulation for the treatment of moderateto-severe Alzheimer’s disease: study protocol for a prospective self-controlled trial
- Ranibizumab versus conbercept for wet age-related macular degeneration: protocol for a prospective cohort study