Mitochondrial oxidative damage and apoptosis induced by high glucose through Rho kinase signal pathway in renal tubular epithelial cells
2018-07-03WenNingLiHuiHanZiYangJingXiaoHongYangYinZhangJiaLiWei
Wen-Ning Li, Hui Han, Zi-Yang Jing, Xiao-Hong Yang, Yin Zhang, Jia-Li Wei
Department of Nephrology, Hainan General Hospital, Haikou Hainan 570311, China
1. Introduction
Diabetic nephropathy (DN) is the major cause of chronic kidney disease and also the primary reason of undergoing kidney replacement therapy[1]. Therefore, to establish a new treatment strategy for DN is of great importance to improve the prognosis of these patients and reduce the socioeconomic burden. Importantly, it is crucial to understand the molecular mechanism of DN.
Over the past decade, a large quantity of evidence have demonstrated that oxidative stress is induced under diabetic conditions[2]. Oxidative stress refers to the overproduction and/ or depletion of active molecules, such as reactive oxygen species(ROS) and reactive nitrogenspecies, resulting in an imbalance between the generation of ROS and antioxidant defenses[3]. Data show that high glucose-induced oxidative stress, particularly the overproduction of mitochondrial ROS in renal tubular epithelial cells, plays a key role in the pathogenesis of DN tubulointerstitial fibrosis. On the one hand, ROS can activate the signal transduction cascade effect, such as PKC, MAPK, JAK/STAT, and activate some transcription -factors including NF-κB, ET-1, TGF-β and ECM.Moreover, these signaling molecules in turn induce ROS production,continually amplifying cellular damage caused by high glucose,eventually leading to progressive glomerulosclerosis and tubular atrophy, and finally causing renal function decline and failure[4].
As an effector of small G protein Rho, Rho kinase has many important physiological and pathological functions, containing cytoskeletal reorganization, cell migration, apoptosis and gene expression[5].Fasudil, targeting the ATP-dependent kinase domain and inhibiting ROCK1 and ROCK2 equivalently, is the most commonly used pharmacological ROCK inhibitor (ROCKi). Fasudil presents a promising approach to the prevention of breast cancer metastasis, heart failure and DN[6-8]. More researches indicated the hypothesis that ROCKi might be nephroprotective in DN. Kikuchi et al[9] has reported that treatment with fasudil prevents the development of diabetes partially by improving adipocyte differentiation in insulin-resistant diabetic rats. It also has beneficial effects on the glomerulosclerosis and interstitial fibrosis by reducing prosclerotic cytokines and ECM expression, and inhibiting epithelial–mesenchymal transition phenotype[10].
At present, the aim of the study was to investigate the potential mechanisms underlying the role of mitochondrial oxidative stress in the pathogenesis of DN. We hypothesized that inhibited Rho kinase could prevent mitochondrial oxidative stress by modulating the Rho kinase pathway in vivo and in vitro.
2. Materials and methods
2.1. Reagents and antibodies
Antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), including rabbit anti-human pro-caspase 3, dynamin-related protein 1 (Drp1), peroxisome proliferator activated receptor gamma coactivator 1 alpha (PGC- 1α), myosin phosphatase target subunit 1(MYPT1) COL1 polyclonal antibodies.
2.2. Cell culture
The immortalized human renal proximal tubular epithelial (HK-2) cell line were purchased from cell bank of Xiangya Central Experiment Laboratory and maintained at 37 ℃ in an atmosphere of 95% air and 5% CO2in RPMI-1640, containing 15% FBS, 1% P/S and 1%Glutamax. After digestion with 0.25% trypsin and 0.02% EDTA, 5×105cells were seeded in six-well plates. Subsequently, the cells were treated with the following protocol: (1) HK-2 cells were treated with 5 mM glucose, 30 mM glucose and 30 mM mannitol for 48 h; (2) HK-2 cells were pre-treated with 10 μmol/L fasudil for 30 min and then cultured with 30 mM glucose for 48 h; (3) HK-2 cells were transfected with ROCK1 siRNA or sc-siRNA before treated with 30 mM glucose.
2.3.Real-time PCR
Total RNA of HK-2 cells were extracted using Trizol Total RNA Isolation kit (Invitrogen). Reverse transcription was performed using the Superscript Ⅲ RT kit (Invitrogen) according to the manufacturer’s instruction. Brie fly, the primer sequences designed by Primer 5 software were synthesised at the Invitrogen Corporation(Shanghai, China) and listed in Table 1. The reactions were incubated as follows: 94 ℃ for 5 min, 94 ℃ 30 s, 59 ℃ 30 s, 72 ℃30 s, 35 cycles, 72 ℃ for 5 min.

Table 1Primers used in this study.
2.4.Western blot analysis
HK-2 cells were lysed in 2×SDS–PAGE sample buffer. Total protein concentrations were measured with a BCA kit (Applygen).Lysate proteins were separated by 10% sodium dodecyl sulfatepolyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane. Then, the membranes were incubated with the following primary antibodies overnight at 4°℃: anti-pro-caspase 3, anti-Drp1,anti-PGC-1α, anti-MYPT1, anti-COL1, beta-actin. Subsequently,the membranes were washed with PBS-Tween and incubated with secondary antibodies for 2 h at room temperature. Immunoreactivity was scanned using an ECL system (Amersham) and the Bio-Rad Electrophoresis Image Analyzer (Bio-Rad, Hemel Hampstead, UK).
2.5. Statistical analysis
Data were expressed as the means ± standard deviation and analyzed using SPSS 20.0 for Windows (SPSS, Chicago, IL, USA).Data were calculated for at least three independent experiments. For comparisons of different groups, Student’s t-test was used, P < 0.05 was considered as statistically significance.
3. Results
3.1. Rho kinase participated in high glucose-induced HK-2 cells
To explore the activity of Rho kinase in high glucose-induced HK-2 cells, we measured the level of MYPT1 which was one of the major substrates of ROCK. In high glucose group, the MYPT1 protein levels were significantly higher than those of 5 mM glucose group and mannitol group (Figure 1).

Figure 1. Upregulation of MYPT1 protein in HK-2 cells induced by high glucose.
3.2. Effects of Rho kinase pathway on mitochondriainduced oxidative damage and apoptosis induced by high glucose in HK-2 cells
HK-2 cells were treated with fasudil at a dose of 10 μM, and ROCK1 siRNAs were successfully transfected into HK-2 cells and then these cells were cultured with 30 mM glucose, seperately.Administration of high glucose for HK-2 cells up-regulated the transcription of manganese superoxide dismutase (MnSOD2) by real-time PCR. Further, treatment with fasudil and ROCK1 siRNA significantly suppressed the increase of high glucose-induced MnSOD2 mRNA expression, respectively (Figure 2). These data suggested that Rho kinase pathway participated in mitochondriainduced oxidative damage induced by high glucose in HK-2 cells.
In addition, our results showed that high glucose treatment upregulated the expression of pro-caspase-3 protein expression.Treatment with fasudil and ROCK1 siRNA significantly antagonized high glucose-induced pro-caspase-3 protein expression, respectively (Figure 3). These data indicated that Rho kinase pathway was involved in mitochondria-related apoptosis induced by high glucose in HK-2 cells.
3.3. Effects of Rho kinase pathway on Drp1 and PGC-1α expression in high glucose-induced HK-2 cells
Mitochondrial morphological changes are controlled by a series of dynamin family GTP-binding proteins, including Drp1. PGC-1α is a regulator of mitochondrial biosynthesis and participates in oxidative phosphorylation. As shown in Figure 4, we measured the expression of Drp1 and PGC-1α in order to explore the mechanism of Rho kinase pathway in high glucose-induced HK-2 cells. In high glucose group, Drp1 and PGC-1α were up-regulated compared with the control group. Importantly, fasudil and ROCK1 siRNA markedly suppressed the expression of Drp1 and PGC-1α compared with high glucose group. Our results suggested that Rho kinase pathway regulated the mitochondrial oxidative damage of high glucoseinduced HK-2 cells by regulating the expression of mitochondrial motor protein Drp1 and mitochondrial gene PGC-1αα.
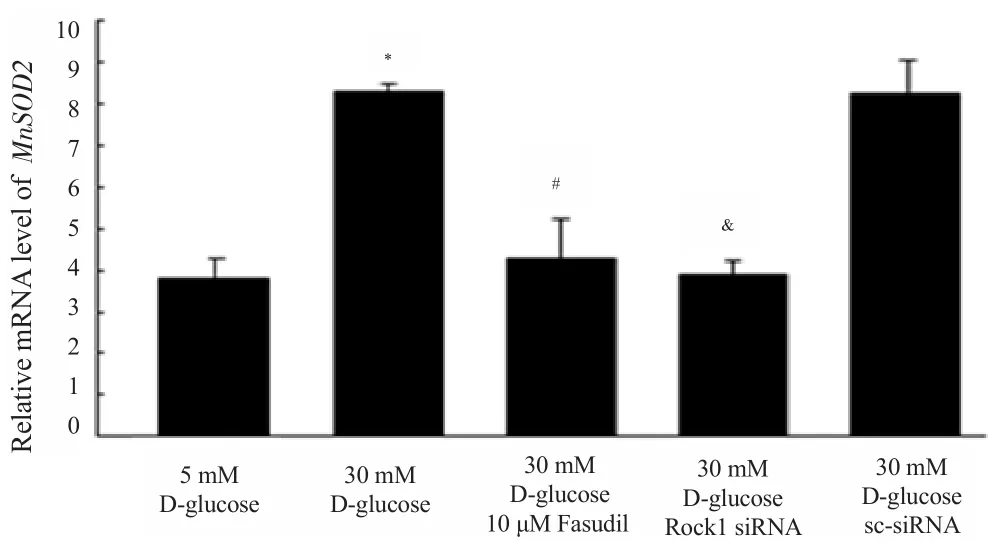
Figure 2. Relative mRNA level of MnSOD2.

Figure 3. Relative protein level of pro-caspase-3.

Figure 4. Mitochondria related proteins levels in high glucose-induced HK-2 cells.
4. Discussion
Our study demonstrated that Rho kinase signaling pathway was involved in the high glucose-induced renal tubular epithelial cell.The Rho kinase inhibitor fasudil and ROCK1 siRNA attenuated the increased levels of MnSOD2 and pro-caspase-3 expression levels in HK-2 cells cultured in high glucose. Rho kinase pathway was involved in mitochondrial oxidative damage and apoptosis of high glucose-reduced renal tubular epithelial cells by regulating Drp1 and PGC-1α expression.
According to previous research, a number of important intracellular signaling pathways were activated in high glucose conditions including the TGF-β/SMAD, nuclear factor-κB and Rho/ROCK[11-13]. They played roles in the inflammation response,RAS activation, epithelial–mesenchymal transition and fibrosis of DN[14,15]. However, we focused on the oxidative damage, in which Rho kinase signaling pathway may serve crucial roles. The production of ROS is closely related to the high glucose-induced oxidative stress in renal tubular epithelium. Most of intracellular ROS comes from mitochondria. Reactive oxygen can cause lipid peroxidation in the mitochondrial membrane, resulting in impaired mitochondrial function, insufficient ATP synthesis, increased free radicals, loss of energy in the cells, increased intracellular calcium concentration, and ultimately lead to apoptosis[16,17]. In this study,we used ROCKi and ROCK1 siRNA to inhibit the activation of Rho kinase pathway. Fasudil competes with ATP for the ATP binding site in the ROCK catalytic domain and blocks ROCK activity through competitive antagonism, thereby blocking the ROCK-mediated biological function[9]. The superoxide dismutase family is a key enzyme in scavenging oxygen free radicals. Among them,MnSOD2 plays a major role, and it is also a marker of oxidative stress[18]. MnSOD2 was significantly up-regulated in high glucose group. More importantly, fasudil and ROCK1 siRNA attenuated the high glucose-induced MnSOD2 mRNA expression, indicating that oxidative stress was inhibited, and mitochondrial oxidative damage to renal tubular epithelial cells was reduced.
Glomerular lesions was traditionally associated with DN. However,evidence has increasingly shown that renal tubule also played a crucial role in DN in recent years[19,20]. Our results are consistent with those results that high glucose-induced oxidative stress and apoptosis are increased in DN[21]. Meanwhile, we observed that the expression of pro-caspase-3, an index for pro-apoptotic processes in vitro, was up-regulated in the renal tubular epithelial cells after exposure to high glucose environment. Excessive ROS could cause destruction of mitochondrial permeability and release of mitochondrial cytochrome C[22], which activated caspase-3 and-9 to induce apoptosis[23]. Moreover, increasing evidence support the role of Rho Rho kinase pathway in DN[24]. Our data show that interference of the ROCK1 expression and Rho kinase activity can reduce high glucose-induced tubular apoptosis. ROCK1 is cleaved by caspase-3 at the C-terminal domain to activate constitutively,and regulate cytoskeleton to induce apoptosis. Taken together,this study demonstrate that Rho kinase pathway may take part in hyperglycemia-induced renal tubular epithelial cell mitochondriamediated apoptosis.
Mitochondria-targeting antioxidants have been suggested as a possible therapeutic strategy to prevent and treat the the development of DN[25,26]. In addition, as in other cells, the Rho signaling pathway is activated in renal cells of the diabetic milieu environment[27]. Studies have demonstrated that the pathway contributes to progrowth, prosclerotic/profibrotic signalling, and increased ECM production[7,28,29]. Furthermore, it mediates the activation of the kidney myofibroblast and participates in the pathophysiological progression of renal fibrosis[30,31]. Indeed,studies showed that ROCK1 was induced by high glucose and then stimulated the endothelial-to-mesenchymal transition[32]. Our data providesevidence that the Rho kinase pathway regulates oxidative damage and apoptosis of high glucose-induced renal tubular epithelial cells by regulating the expression of Drp1 and PGC-1α.More importantly, the present study suggests that mitochondrial oxidative stress regulated by Rho kinase pathway plays a key role in renal tubulointerstitial fibrosis in DN. Further studies using gene knockout animal models and more clinical trials using antioxidants will be required to elucidate the distinct roles of mitochondrial oxidative stress in renal tubular epithelial cells in DN.
In summary, our evidence shows that Rho kinase pathway is involved in mitochondrial oxidative stress of high glucose-reduced renal tubular epithelial cells by regulating Drp1 and PGC-1α. This study provides new insights into the pathogenesis of DN and it should help develop a new therapeutic strategy for DN.
Conflict of interest statement
The authors declare that they have no Conflict of interest.
[1] Shen Z, Fang Y, Xing T, Wang F. Diabetic nephropathy: From pathophysiology to treatment. J Diabetes Res 2017; 2017: 2379432.
[2] Sagoo MK, Gnudi L. Diabetic nephropathy. Is there a role for oxidative stress? Free Radic Biol Med 2018; 116: 50-63.
[3] Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001; 414(6865): 813-820.
[4] Lee HB, Yu MR, Yang Y, Jiang Z, Ha H. Reactive oxygen speciesregulated signaling pathways in diabetic nephropathy. J Am Socnephrol 2003; 14: S241-S245.
[5] Fukata Y, Amano M, Kaibuchi K. Rho-Rho-kinase pathway in smooth muscle contraction and cytoskeletal reorganization of non-muscle cells.Trends Pharmacol Sci 2001; 22(1): 32-39.
[6] Guerra FS, Oliveira RGD, Fraga CAM, Mermelstein CDS, Fernandes PD.ROCK inhibition with Fasudil induces beta-catenin nuclear translocation and inhibits cell migration of MDA-MB 231 human breast cancer cells.Sci Rep 2017; 7(1): 13723.
[7] Kolavennu V, Zeng L, Peng H, Wang Y, Danesh FR. Targeting of RhoA/ROCK signaling ameliorates progression of diabetic nephropathy independent of glucose control. Diabetes 2008; 57(3): 714-723.
[8] Guan P, Liang Y, Wang N. Fasudil alleviates pressure overload-induced heart failure by activating Nrf2-mediated antioxidant responses. J Cell Biochem 2018; doi: 10.1002/jcb.26662.
[9] Kikuchi Y, Yamada M, Imakiire T, Kushiyama T, Higashi K, Hyodo N, et al. A Rho-kinase inhibitor, fasudil, prevents development of diabetes and nephropathy in insulin-resistant diabetic rats. J Endocrinol 2007; 192(3):595-603.
[10] Komers R, Oyama TT, Beard DR, Tikellis C, Xu B, Lotspeich DF, et al. Rho kinase inhibition protects kidneys from diabetic nephropathy without reducing blood pressure. Kidney Int 2011; 79(4): 432-442.
[11] Tang SC, Leung JC, Lai KN. Diabetic tubulopathy: An emerging entity.Contrib Nephrol 2011; 170: 124-134.
[12] Kanlaya R, Sintiprungrat K, Thongboonkerd V. Secreted products of macrophages exposed to calcium oxalate crystals induce epithelial mesenchymal transition of renal tubular cells via RhoA-dependent TGF-beta1 pathway. Cell Biochem Biophys 2013; 67(3): 1207-1215.
[13] Manickam N, Patel M, Griendling KK, Gorin Y, Barnes JL. RhoA/Rho kinase mediates TGF-beta1-induced kidney myofibroblast activation through Poldip2/Nox4-derived reactive oxygen species. Am J Physiol Renal Physiol 2014; 307(2): F159-F171.
[14] Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensinconverting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med 1993; 329(20): 1456-1462.
[15] Liu Y. Epithelial to mesenchymal transition in renal fibrogenesis:Pathologic significance, molecular mechanism, and therapeutic intervention. J Am Soc Nephrol 2004; 15(10): 1-12.
[16] Viola HM, Hool LC. Targeting calcium and the mitochondria in prevention of pathology in the heart. Curr Drug Targets 2011; 12(5): 748-760.
[17] Loor G, Kondapalli J, Iwase H, Chandel NS, Waypa GB, Guzy RD, et al.Mitochondrial oxidant stress triggers cell death in simulated ischemia–reperfusion. Biochim Biophys Acta 2011; 1813(7): 1382-1394.
[18] Bresciani G, Da CI, Gonzalez-Gallego J. Manganese superoxide dismutase and oxidative stress modulation. Adv Clin Chem 2015; 68: 87-130.
[19] Hills CE, Squires PE. The role of TGF-beta and epithelial-to mesenchymal transition in diabetic nephropathy. Cytokine Growth Factor Rev 2011; 22: 131-139.
[20] Wang JY, Yang JH, Xu J, Jia JY, Zhang XR, Yue XD, et al. Renal tubular damage may contribute more to acute hyperglycemia induced kidney injury in non-diabetic conscious rats. J Diabetes Complications 2015;29(5): 621-628.
[21] Terami N, Ogawa D, Tachibana H, Hatanaka T, Wada J, Nakatsuka A, et al. Long-term treatment with the sodium glucose cotransporter 2 inhibitor, dapagliflozin, ameliorates glucose homeostasis and diabetic nephropathy in db/db mice. Plos One 2014; 9(6): e100777.
[22] Orsini F, Migliaccio E, Moroni M, Contursi C, Raker VA, Piccini D, et al. The life span determinant p66Shc localizes to mitochondria where it associates with mitochondrial heat shock protein 70 and regulates transmembrane potential. J Biol Chem 2004; 279(24): 25689-25695.
[23] Susztak K, Raff AC, Schiffer M, Bottinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes 2006; 55(1): 225-233.
[24] Matoba K, Kawanami D, Nagai Y, Takeda Y, Akamine T, Ishizawa S,et al. Rho-Kinase blockade attenuates podocyte apoptosis by inhibiting the notch signaling pathway in diabetic nephropathy. Int J Mol Sci 2017;18(8): pii: E1795. doi: 10.3390/ijms18081795.
[25] Lambooy SPH, Bidadkosh A, Nakladal D, van Buiten A, Girgis RAT, van der Graaf AC, et al. The novel compound sul-121 preserves endothelial function and inhibits progression of kidney damage in type 2 diabetes mellitus in mice. Sci Rep 2017; 7(1): 11165.
[26] Galvan DL, Badal SS, Long J, Chang BH, Schumacker PT, Overbeek PA, et al. Real-time in vivo mitochondrial redox assessment confirms enhanced mitochondrial reactive oxygen species in diabetic nephropathy.Kidney Int 2017; 92(5): 1282-1287.
[27] Komers R. Rho kinase inhibition in diabetic kidney disease. Br J Clin Pharmacol 2013; 76(4): 551-559.
[28] Peng F, Wu D, Gao B, Ingram AJ, Zhang B, Chorneyko K, et al. RhoA/Rho-kinase contribute to the pathogenesis of diabetic renal disease.Diabetes 2008; 57(6): 1683-1692.
[29] David M, Petit D, Bertoglio J. Cell cycle regulation of Rho signaling pathways. Cell Cycle 2012; 11(16): 3003-3010.
[30] Nagatoya K, Moriyama T, Kawada N, Takeji M, Oseto S, Murozono T,et al. Y-27632 prevents tubulointerstitial fibrosis in mouse kidneys with unilateral ureteral obstruction. Kidney Int 2002; 61(5): 1684-1695.
[31] Manickam N, Patel M, Griendling KK, Gorin Y, Barnes JL. RhoA/Rho kinase mediates TGF-beta1-induced kidney myofibroblast activation through Poldip2/Nox4-derived reactive oxygen species. Am J Physiol Renal Physiol 2014; 307(2): F159-F171.
[32] Galvan DL, Badal SS, Long J, Chang BH, Schumacker PT, Overbeek PA, et al. Real-time in vivo mitochondrial redox assessment confirms enhanced mitochondrial reactive oxygen species in diabetic nephropathy.Kidney Int 2017; 92(5): 1282-1287.
杂志排行

Asian Pacific Journal of Tropical Medicine的其它文章
- Traditional medicines and their in-vitro proof against Staphylococcus aureus in Pakistan
- Interferon-induced protein with tetratricopeptide repeats 1 (IFIT1)polymorphism as a genetic marker of cerebral malaria in Thai population
- Elevated serum nitric oxide and hydrogen peroxide levels as potential valuable predictors of herpes zoster
- Prevalence and antimicrobial resistance of non-typhoid Salmonella in military personnel, 1988-2013
- Preventive effect of Angelica gigas Nakai extract oral administration on dry eye syndrome
- Potential effect of Silybum marianum L. and Cistus ladaniferus L.extracts on urine volume, creatinine clearance and renal function