基于COI基因序列片段的柔突叶蝉属部分近缘种分子鉴定与系统发育
2018-06-22姚亚林陈祥盛
姚亚林,陈祥盛,杨 琳*
(1.贵州大学 林学院,贵州 贵阳 550025;2.贵州大学 昆虫资源开发利用省级特色重点实验室,贵州 贵阳 550025;3.贵州大学 昆虫研究所,贵州 贵阳 550025;4.贵州大学 贵州山地农业病虫害省级重点实验室,贵州 贵阳 550025)
叶蝉类昆虫均属植食性,不仅刺吸危害植物,而且其刺吸式口器刺吸植物汁液,影响植物的正常发育;取食产卵过程造成的植物伤痕影响水分和养分运输;通过交叉寄主传播植物病毒;排泄物蜜露堵塞植物气孔影响光合作用;分泌物可为多种霉菌提供培养基导致霉病爆发,是农、林、果树及经济植物的重要害虫类群之一。
柔突叶蝉属AbrusDaietZhang,2002属半翅目Hemiptera,叶蝉科Cicadellide,角顶叶蝉亚科Deltocephalinae。该属目前已知19种[1],分别为:安龙柔突叶蝉A.anlongensisChen,Yang Li,2012;竹柔突叶蝉A.bambusanusChen,Yang Li,2012;叉茎柔突叶蝉A.bifurcatusDai Zhang,2002;双枝柔突叶蝉A.biprocessusLi,2011;短板柔突叶蝉A.breviolusDai Zhang,2008;短茎柔突叶蝉A.brevisDai Zhang,2002;凹板柔突叶蝉A.concavelusLi Wang,2006;锥尾柔突叶蝉A.coneusDai Zhang,2002;大明山柔突叶蝉A.damingshanensisXing Li,2014;道真柔突叶蝉A.daozhenensisChen,Yang Li,2012;宽顶侧突柔突叶蝉A.expansivusXing Li,2014;细茎柔突叶蝉A.graciaedeagusLi,2011;衡山柔突叶蝉A.hengshanensisDai Zhang,2002;黄氏柔突叶蝉A.huangiDai Zhang,2002;崀山柔突叶蝉A.langshanensisYang & Chen,2013;雷公山柔突叶蝉A.leigongshanesisLi Wang,2006;武夷柔突叶蝉A.wuyiensisDai Zhang,2002;习水柔突叶蝉A.xishuiensisYang Chen,2013;云山柔突叶蝉A.yunshanensisChen, Yang Li,2012分布于东洋界和古北界。除叉茎柔突叶蝉大明山柔突叶蝉宽顶侧突柔突叶蝉和武夷柔突叶蝉4个种外,其余14种均报道危害竹子。该属形态结构相似,且部分种的雄性外生殖器结构差异程度不大,这对该类昆虫的物种鉴定带来了困难。
目前,研究近似种最有效的方法是结合形态和分子数据进行分类鉴定。关于叶蝉科的分子鉴定方面,一些类群如角顶叶蝉类Deltocephalus-like,弯钩叶蝉属Flexamia,黄翅叶蝉属Dalbulus的16S rRNA基因序列片段可用于区分近缘种[2-3],然而有些类群如缅甸安小叶蝉Anakaburmensis,COI基因也具有相当的鉴定功效[4-5],可见不同动物类群的最适条形码并非一致。在叶蝉类昆虫的DNA条形码研究中,相比于COI基因,16S rRNA基因序列片段较易扩增,常被作为一种有效的分子标记,用于区分形态近似种或描述新发现的种类[6-10],然而,柔突叶蝉属的分子鉴定相关研究尚未有报道。为此,本文提取扩增了采自竹类的柔突叶蝉属5个近似种(锥尾柔突叶蝉、道真柔突叶蝉、习水柔突叶蝉、安龙柔突叶蝉和短茎柔突叶蝉,图A1-E5)COI序列片段,对其进行分子鉴定,同时利用邻接法(NJ)、最大简约MP法构建系统发育树,并结合形态结构与系统发育树对其近缘种的物种分化时间估计进行分析和讨论,以期为角顶叶蝉亚科的分类学和系统学提供科学依据。
1 材料与方法
1.1 实验材料
本研究所用标本的采集信息详见表1,锥尾柔突叶蝉、道真柔突叶蝉、习水柔突叶蝉、安龙柔突叶蝉和短茎柔突叶蝉的外部形态特征及雄性外生殖器如图1、图2。样品冷冻保存于贵州大学昆虫研究所,雄性外生殖器保存于甘油中,以备查用。5种近缘柔突叶蝉的外部形态[11]如图1,其雄性阳茎如图2。
1.2 试剂及仪器
昆虫DNA提取试剂盒购于Omega BioTek公司(D0926-01);引物由生物工程(上海)股份有限公司合成。Olympus SZ2-ILST型光学体视显微镜(德国Leica公司);T100TMThermal Cycler型PCR扩增仪(美国BIO-RAD公司);170-8170 型 UVP 凝胶成像系统(美国 BIO-RAD 公司);Power PacTMHV Power Supply型电泳仪(美国BIO-RAD公司);Mini-10K微型高速离心机(珠海黑马医学仪器有限公司);HVE-50型自动高压灭菌器(日本HIRAYAMA制造公司)等。

表1 供试昆虫的采集信息Tab.1 Collecting information for the insect specimens

A1- A3:安龙柔突叶蝉A.anlongensis;B1- B3:短茎柔突叶蝉A.brecis;C1- C3:锥尾柔突叶蝉A.coneus;D1- D3:习水柔突叶蝉A.xishuiensis;E1- E3:道真柔突叶蝉A.daozhenensis
图1 柔突叶蝉属5个近似种成虫背面、颜面及头胸侧面观
Fig. 1 Habitus of five similar species ofAbrus
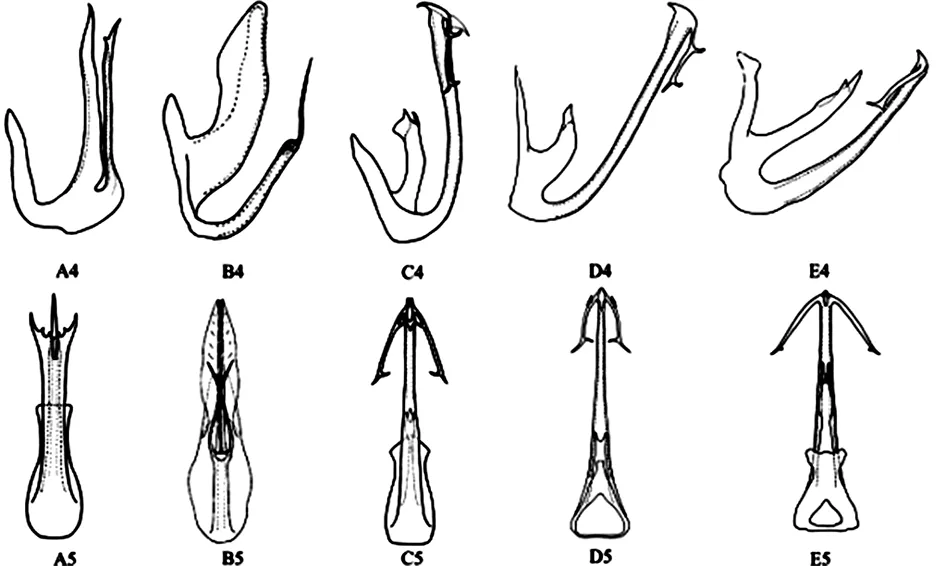
A4- A5:安龙柔突叶蝉A.anlongensis;B4- B5:短茎柔突叶蝉A.brecis;C4- C5:锥尾柔突叶蝉A.coneus;D4- D5:习水柔突叶蝉A.xishuiensis;E4- E5:道真柔突叶蝉A.daozhenensis
图2 柔突叶蝉属5个近似种雄性外生殖器特征
Fig. 2 Morphology of male external genitals of five similar species ofAbrus
1.3 DNA提取及PCR扩增
冷冻保存标本解冻后,将腹部放入甘油中保存,剩余的虫体取头胸足等部分研磨至粉末状,然后用Omega BioTek公司生产的E.Z.N.A.TMInsect DNA kit试剂盒提取总DNA,产物-20℃保存。
PCR上下游扩增引物参照Folmer等[5]设计,COI片段引物为:5′-GGTCAACAAATCATAAAGATATTG-3′;5′-TAAACTTCAGGGTGACCAAAAAAT-3′。PCR反应体系为30 μL,其中模板DNA 3 μL,上下游引物各1 μL、Taq PCR Master Mix 15 μL、ddH2O补至30 μL。PCR扩增程序:94℃预变性3 min,94℃变性30 s,50℃退火30 s,72℃延伸1 min,33个循环,最后72℃补偿延伸10 min,4℃保存。PCR产物取,3 μL采用1%琼脂糖凝胶电泳检测,纯化步骤参照DNA凝胶回收试剂盒Omega BioTek公司(D0926-01)程序。回收产物送至生工生物工程(上海)股份有限公司进行双向测序。
1.4 数据分析
将获得的测序峰图利用DNAstar 5.0软件进行正反链校对和编辑,手动去除序列两端的引物区,获得有效片段的样本序列。Meglign进行排序后利用Clustal X进行多序列比对,采用DnaSP v5计算信息位点、变异位点数,通过MEGA 6.06分析碱基组成、变异位点,并基于Kimura-2-parameter(K2P)模型进行遗传距离分析,并以叉突平额叶蝉Flatfrontapronga为外群,采用邻接法(Neighbor-Joining,NJ法)和最大简约法(MP法)构建系统发育树,系统发育树的节点支持率采用自展值进行估计,重复检验1000次。同时结合贝叶斯方法BPP[12-15]和MCMCTREE[16]对这5个近缘种的物种分化时间进行讨论。
2 结果与分析
2.1 碱基组成及序列变异
利用MEGA 6. 06软件对本研究中获得的COI基因序列进行比对后,保留622 bp的同源性序列,对其分析结果显示:COI序列中,A、T、G、C含量分别是30.5% 、36.8% 、14.7% 、18.0%,A+ T的平均含量为67.3%,G + C的平均含量为32.7%;共检测到保守位点407个,变异位点215个,简约信息点90个,自裔位点125个,变异位点约占总序列的34.56%。
2.2 遗传距离
利用MEGA 6.06软件计算不同个体的COI基因序列间的遗传距离(不同模型下的遗传距离,表3) 。习水柔突叶蝉与道真柔突叶蝉种间的遗传距离最小为0.011,锥尾柔突叶蝉与道真柔突叶蝉遗传距离最大为0.095。柔突叶蝉属与平额叶蝉属间的遗传距离为0.69~0.96。从不同模型下的遗传距离 (表3~7) 可以看出,K2P转换+G遗传距离与Pdistance+U遗传距离模型下短茎柔突叶蝉与安龙柔突叶蝉种间遗传距离最小为0.011,最大为0.103~0.335,此时从遗传距离界定这两种较困难。锥尾柔突叶蝉和道真柔突叶蝉间遗传距离在K2P转换+颠换+G模型下最大,为0.367,而在JC69 +Gamma模型下为0.366,Pdistance+U遗传距离最小为0.095。表明转换与颠换以及位点的替代速率分布的异质性较强,对遗传距离的估计影响较大,利用COI基因序列间遗传差异来鉴定柔突叶蝉效率不太高,总体来说,COI基因序列可以暂时被作为该属近缘种分子鉴定的候选基因序列片段。

表3 供试5种柔突叶蝉COI基因序列的Pdistance遗传距离Tab.3 Genetic distances under the Pdistance model of COI sequence of five Abrus species
注:右上半三角部分为遗传距离的标准误S.E,下同。
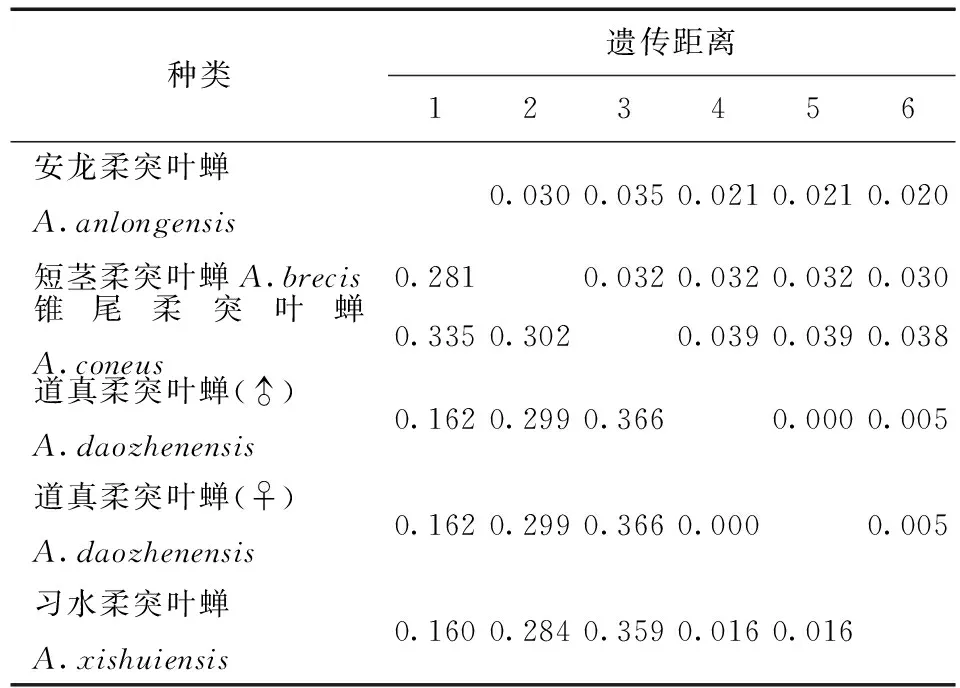
表4 供试5种柔突叶蝉COI基因序列的JC69+G遗传距离Tab.4 Genetic distances under the JC69+U model of COI sequence of five Abrus species
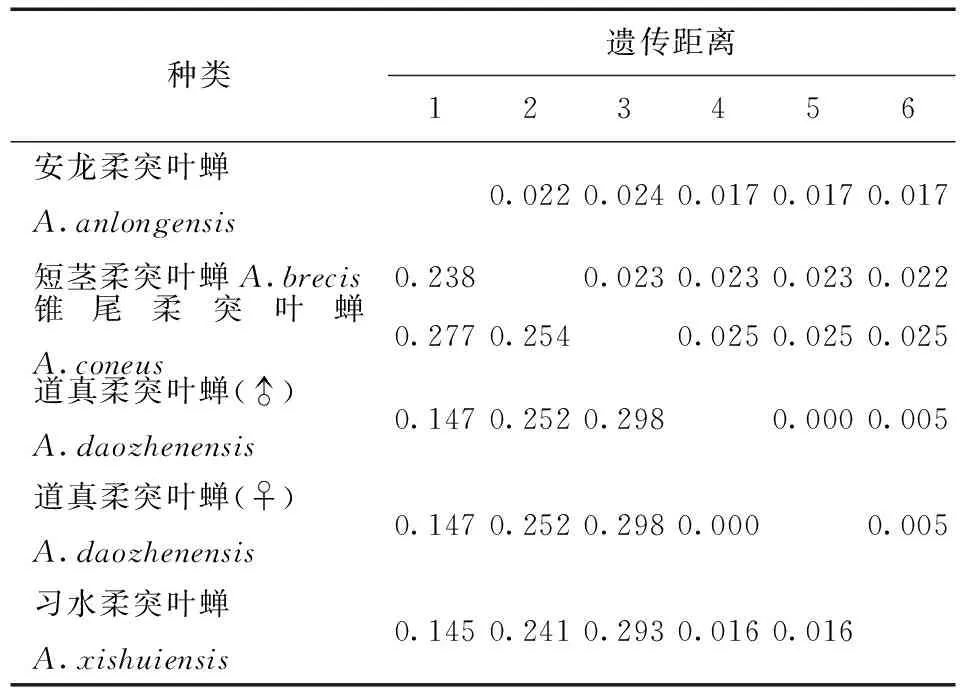
表5 供试5种柔突叶蝉COI基因序列的JC69+U遗传距离Tab.5 Genetic distances under the K2P+Ts+Tv+G model of COI sequence of five Abrus species
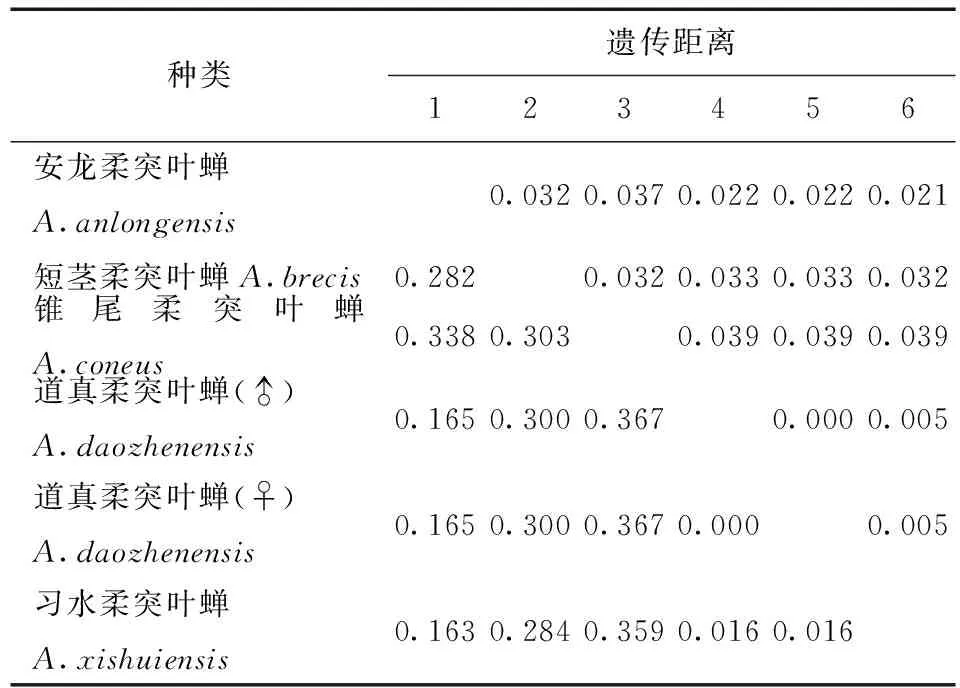
表6 供试5种柔突叶蝉COI基因序列的K2P转换+颠换+G遗传距离Tab.6 Genetic distances under the K2P+Ts +G model of COI sequence of five Abrus species
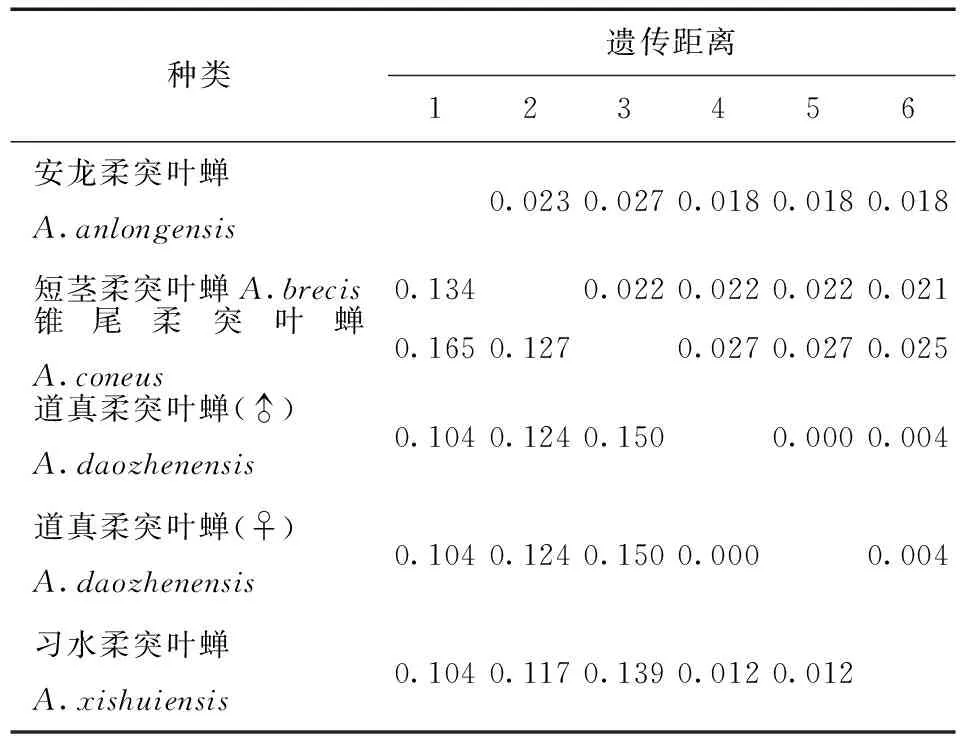
表7 供试5种柔突叶蝉COI基因序列的K2P转换+G遗传距离Tab.7 Genetic distances under the K2P+Ts +G model of COI sequence of five Abrus species

表8 供试5种柔突叶蝉COI基因序列的K2P转换+U遗传距离Tab.8 Genetic distances under the K2P+Ts +U model of COI sequence of five Abrus species
2.3 碱基替换饱和度分析
本研究使用F84遗传距离对数据组进行了替换饱和分析,结果表明,转换(S)与颠换(V)的比率均随着遗传距离的增加而上升,颠换逐渐多于转换,与线粒体基因中碱基CT及AG突变较多的情况相吻合。转换与颠换的曲线均呈现出向平台期过渡的趋势,即出现饱和状态的倾向。整体而言,尽管序列整体上较为保守,但构建的数据集饱和度在较低的水平上,说明作为常用的分子标记,COI基因包含足够的遗传信息可以用于解析本研究所构建数据集的分类关系(图3)。

表9 供试5种柔突叶蝉COI基因序列的Pdistance+U遗传距离Tab.9 Genetic distances under the Pdistance+U model of COI sequence of five Abrus species
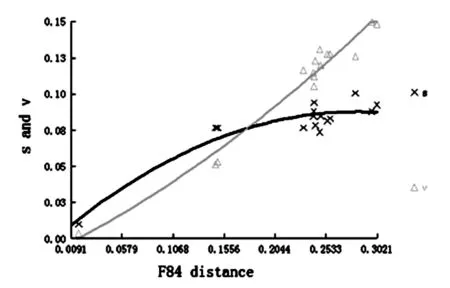
横坐标:F84模型下的遗传距离;纵坐标:转换和颠换值
图3 序列碱基替换饱和度分析
Fig.3 Transitions (s) and transversions (v) versus divergences by COI sequences.
2.4 系统发育分析
以叉突平额叶蝉为外群,基于COI构建的系统发育树中(图4、5),得到了一致的拓扑结构,并得到了较高的检验支持率。道真柔突叶蝉和习水柔突叶蝉的3个种聚为一支,与安龙柔突叶蝉形成姐妹群关系,短茎柔突叶蝉和锥尾柔突叶蝉位于较根部,其中道真柔突叶蝉与习水柔突叶蝉亲缘关系最近,而其与锥尾柔突叶蝉和短茎柔突叶蝉亲缘关系较远。基于贝叶斯方法的BPP物种定界与物种树估计的结果(图6)也表明锥尾柔突叶蝉与道真柔突叶蝉的亲缘关系远,尽管它们的形态极为相近,但COI构建的系统发育树中并未按属聚一簇,其单系性并不明确。
MCMCTREE的估计结果中(图7),短茎柔突叶蝉、锥尾柔突叶蝉、道真柔突叶蝉与习水柔突叶蝉三种在系统树中的关系与BPP的一致,但其三物种的最近共同祖先(most recent common ancestor,MRCA)分化时间比BPP估计的高,道真柔突叶蝉与习水柔突叶蝉的分化时间约为4-10ka BP左右,而锥尾柔突叶蝉和道真柔突叶蝉+习水柔突叶蝉+安龙柔突叶蝉+锥尾柔突叶蝉的MRCA分化时间约120 ka BP,都是处于全新世初,冰后期。锥尾柔突叶蝉位于根部,这也与其常见寄主差异有关,锥尾柔突叶蝉常见的寄主并非方竹,而是平竹,故在系统发育树中体现出与其他柔突叶蝉的差异而出现在根部。
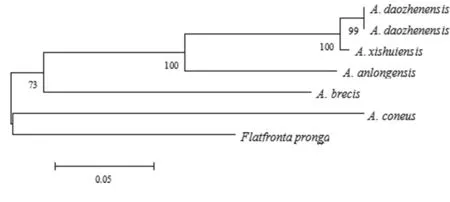
图4 Abrus 5个近缘种的COI 基因序列进化树(NJ tree)
Fig.4 eighbor-joining (NJ) analysis of Kimura-2-parameter (K2P) distances of fiveAbrusspecies. Values above the branches are NJ bootstrap values (> 60%).
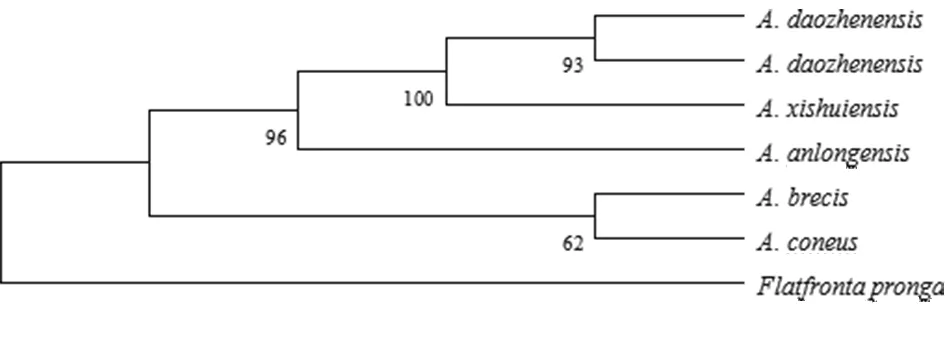
图5 Abrus 5个近缘种的COI基因序列进化树(MP tree)
Fig.5 Strict consensus phylogenetic tree based on maximum parsimony (MP) analysis of fiveAbrusspecies. Values above the branches are MP bootstrap values (> 60%).
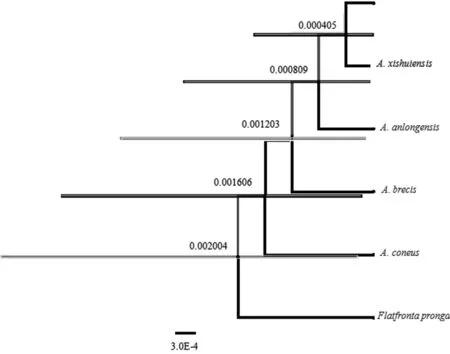
图6 Abrus 5个近缘种的BPP物种定界
与物种树估计的最优结果
Fig.6 The optimal results of species delimitation and species tree estimation by BPP of fiveAbrusspecies

图7 Abrus 5个近缘种的MCMCTREE物种分化时间估计
Fig.7 Species divergence time of the fiveAbrusspecies by MCMCTREE software
3 结论与讨论
目前已知柔突叶蝉Abrus共19种,全部分布于中国,其形态相似度高,且大部分寄主为竹类。本研究的5种柔突叶蝉的寄主有贵州都匀斗篷山特有的毛环方竹Chimonobambusahirtinoda分布,习水的方竹Chimonobambusaangustifolia以及宽阔水有分布的平竹Qiongzhueacommunis和一些其他的方竹,由寄主的独有性以及柔突叶蝉在世界分布来看,尤其是该属与方竹共有特有区的分布特点来看,该属似乎与竹类的进化有较强的协同性,但其实际协同关系还待深入研究。
依据经典分类方法利用雄性外生殖器的形态学特征对这5种柔突叶蝉进行鉴定时,锥尾柔突叶蝉(C1~C3)与习水柔突叶蝉(D1~D3)道真柔突叶蝉(E1~E3)三者的雄性外生殖器也极为相似(表2),使得在农林生产实践中基于形态学来区分这三物种较困难。本研究首次利用COI序列进行分子鉴定,从遗传距离来看,COI做分子鉴定是可行的,但在不同模型下的距离差异较大可能预示着当样本数量增大可能表现并不可观,今后应该结合其他基因片段评价其鉴定效率。同时,COI的系统发育结果表明锥尾柔突叶蝉和道真柔突叶蝉与习水柔突叶蝉存在很大差异。从形态来看,其主要区别不仅在于其翅的外观色泽、颜面额唇基的饱满度等特征等,而雄性外生殖器的连锁形态、阳茎端部的反向齿突也明显不同,COI却能很好的体现这些隐存的差异,Fang等[17]提出“Y”连锁更具祖征的结论,本文中锥尾柔突叶蝉和道真柔突叶蝉及习水柔突叶蝉的连锁却差异较大,前者是Abrus中唯一一个主干较细的,其余均较粗大(表1),因此出现COI的差异表现并不意外。
分子鉴定理想的条形码种内遗传差异会小于近缘物种种间差异[18],即种内、种间遗传差异的幅度确定了物种的界限[19]。若种内、种间遗传差异存在重叠,则说明该分子标记无法准确区分物种。理论上,有效的DNA条形码应存在条形码间隔,即种内、种间遗传距离差异明显。Hebert等[20]提出,种间遗传差异应达到种内遗传差异的10倍。本研究所涉及种类的COI基因片段序列均有明显条形码间隔,未出现种内、种间遗传差异存在重叠的种群,证实了它们作为慈竹叶蝉类昆虫DNA条形码标准基因的可行性。尽管有研究表明角顶叶蝉类Deltocephalus-like,弯钩叶蝉属Flexamia,黄翅叶蝉属Dalbulus 16S rRNA基因序列片段可用于区分近缘种[2-3]。但本文研究结果表明至少竹类柔突叶蝉的COI基因序列片段被作为一种有效的分子标记,用于区分形态近似种。
COI基因的普遍替代速率从0.0168到0.023每个位点/每百万年[21-22]。真核生物的替代速率从0.00172到0.0258每个位点/每百万年[23-24]。昆虫的16S或12S-16S的替代速率从0.005到0.011[22,25]。本研究使用COI的替代速率0.0168到0.023。从MCMCTREE估计的结果来看(图7),短茎柔突叶蝉、锥尾柔突叶蝉、道真柔突叶蝉与习水柔突叶蝉的分化时间约为4ka BP左右,而锥尾柔突叶蝉与道真柔突叶蝉与习水柔突叶蝉的祖先分化时间约16ka BP,都是处于全新世初,冰后期,但是估计结果偏低,这很可能与这些物种的分化早期出现过水平基因流或基因渐渗等复杂演历程有关,而本文选择的COI突变频率的先验分布尽管是信息较少的先验,但是数据量少使得估计结果精度低。全新世至近期经历了多次的冷暖波动[26],植被的垂直带谱逐渐完善的过程中,一些特殊的植物北移,如水青冈等,箭竹等高海拔植物下移[27],而宽阔水国家级自然保护区正好拥有水青冈-箭竹林群落,同时也具有锥尾柔突叶蝉与道真柔突叶蝉两种柔突叶蝉昆虫的分布,这两种柔突叶蝉的生殖器明显和贵州雷公山的安龙柔突叶蝉,以及贵州都匀斗篷山的短颈柔突叶蝉的阳茎不同,而这三个地方的方竹自冰后期全新世以来,在经历分化并形成当前的区系分布格局的进程中,应该对柔突叶蝉属的分布格局产生重大影响,如果这推断成立,那么从这种若隐若现而有趣的寄主-昆虫多样性联系中可以深入分析柔突叶蝉类群的演化及协调进化机制,不过今后还需进一步结合该属的更多的地理种群样本从谱系地理学来研究探讨更明晰的结论。
参 考 文 献:
[1] Zahniser N. 2007-present. An online interactive key and searchable database of Deltocephalinae (Hemiptera: Cicadellidae). http://zahniser.speciesfile.org/
[2] Fang Q, Black C, Blocker D,etal. A phylogeny of New World Deltocephalus-like leafhopper genera based on mitochondrial 16S ribosomal DNA sequences [J].MolecularPhylogeneticsandEvolution, 1993,2 (2):119-131.
[3] Dietrich C, Whitcomb R, Black C. Phylogeny of the grassland leafhopper genus Flexamia (Homoptera: Cicadellidae) based on mitochondrial DNA sequences [J].MolecularPhylogeneticsandEvolution, 1997(8):139-149.
[4] Gaunt W, Miles A. An insect molecular clock dates the origin of the insects and accords with palaeontological and biogeographic landmarks [J].MolecularBiologyandEvolution, 2002,19(5):748-761.
[5] 周宁宁. 基于线粒体 CO I和COⅡ基因的茶园假眼小绿叶蝉地理种群的遗传分化研究[D]. 杭州:中国计量学院,2014.
[6] Schuchert P. The European athecate hydroids and their medusae (Hydrozoa, Cnidaria): Capitata Part 1 [M]. Revue Suisse de Zoologie, 2006(113):325-410.
[7] Miglietta P, Schuchert P, Cunningham W. Reconciling genealogical and morphological species in a worldwide study of the family Hydractiniidae (Cnidaria, Hydrozoa) [J].ZoologicaScripta, 2009(38):403-430.
[8] Miranda S, Collins G, Marques C. Molecules clarify a Cnidarian life cycle﹣the “Hydrozoan” Microhydrula limopsicola is an early life stage of the Staurozoan Haliclystus antarcticus[J].PLoSONE, 2010(5):e10182.DOI: 10.1371/journal.pone.0010182.
[9] Moura J, Cunha R, Porteiro M,etal. Polyphyly and cryptic diversity in the hydrozoan families Lafoeidae and Hebellidae (Cnidaria: Hydrozoa) [J].InvertebrateSystematics, 2011a(25):454-470.
[10] Moura J, Cunha R, Porteiro M,etal. The use of the DNA barcode gene 16S mRNA for the clarification of taxonomic problems within the family Sertulariidae (Cnidaria, Hydrozoa) [J].ZoologicaScripta, 2011b(40):520-537.
[11] 陈祥盛,杨琳,李子忠. 中国竹子叶蝉[M]. 北京:中国林业出版社,2012.
[12] Yang Z, Rannala B. Bayesian species delimitation using multilocus sequence data [J].ProceedingsoftheRoyalSocietyB-BiologicalSciences,USA, 2010(107):9264-9269.
[13] Rannala B, Yang Z. Improved reversible jump algorithms for Bayesian species delimitation [J].Genetics, 2013(194):245-253.
[14] Yang Z, Rannala B. Unguided species delimitation using DNA sequence data from multiple loci [J].MolecularBiologyandEvolution, 2014(31):3125-3135.
[15] Rannala B, Yang Z. Efficient Bayesian species tree inference under the multispecies coalescent [J].SystermaticBiology, 2017(66):823-842.
[16] Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Molecular Biology and Evolution, 2007,24(8):1586-91.
[17] Fang Q, Black C, Bbcker D. Cladistic Analysis of Nearctic Deltocephalus-Like Leafhoppers (Homoptera: Cicadellidea) Using Morphological and Molecular Data [J].AnnualsofEntomologySocietyofAmerica, 1995,88(5): 316-323.
[18] Del-Prado R, Cubas P, Lumbsch T,etal. Genetic distances within and among species in monophyletic lineages of Parmeliceae (Ascomycota) as a tool for taxon delimitation [J].MolecularPhylogeneticsandEvolution, 2010(56):125-133.
[19] K?倠hler F. From DNA taxonomy to barcoding: how a vague idea evolved into a biosystematic tool [J].MitteilungenausdemMuseumfürNaturkundeBerlinZoologischeReihe, 2007(83):44-51.
[20] Hebert N, Ratnasingham S, Waard R. Barcoding animal life: cytochrome c oxidase subunit I divergences among closely related species [J].ProceedingsoftheRoyalSocietyB-BiologicalSciences,USA, 2003(270):S96-S99.
[21] Allegrucci G, Trucchi E, Sbordoni V. Tempo and mode of species diversification in Dolichopoda cave crickets (Orthoptera, Rhaphidophoridae) [J].MolecularPhylogeneticsandEvolution. 2011,60(1): 108-121
[22] Papadopoulou A, Anastasiou I, Vogler P. Revisiting the Insect Mitochondrial Molecular Clock: The Mid-Aegean Trench Calibration [J].MolecularBiologyandEvolution, 2010,27(7): 1659-1672.
[23] Kasuga T, White J, Taylor W. Estimation of nucleotide substitution rates in eurotiomycete fungi [J].MolecularBiologyandEvolution, 2001,19(12): 2318-2324.
[24] Percy M, Kiisiowicz R, Panzera F,etal. Origin and phylogeography of the Chagas disease main vector Triatoma infestans base on nuclear rDNA sequences and genome size[J].InfectionGeneticsandEvolution, 2006(1):46-62.
[25] Brower Z. Rapid Morphological Radiation and Convergence among Races of the Butterfly Heliconius-Erato Inferred from Patterns of Mitochondrial-DNA Evolution [J].ProceedingsoftheRoyalSocietyB-BiologicalSciences,USA, 1994,91(14):6491-6495.
[26] 方修琦,侯光良. 中国全新世气温序列的集成重建[J]. 地理科学, 2011,31(4):386-691.
[27] 吕伊娜,熊康宁,容 丽,等. 梵净山生物生态演化的世界自然遗产价值对比分析[J]. 世界地理研究, 2016,25(5):131-141.
