高糖对肾小球系膜细胞BKCa-Orai1信号复合物表达与功能的影响
2018-06-08闫德俊杨云云
杜 鹃,闫德俊,杨云云,陈 硕,魏 圆,沈 兵
(安徽医科大学基础医学院生理学教研室,安徽 合肥 230032)
糖尿病肾病(diabetic nephropathy,DN)是糖尿病最严重的并发症之一,也是糖尿病患者死亡的重要原因[1-2]。肾血流动力学异常是DN早期的重要特点,表现为肾小球高压、高灌注和高滤过,这些异常情况长期存在使肾小球系膜基质及基底膜合成增加、降解减少,引起肾小球硬化,最终导致DN的发生[3]。肾小球系膜细胞(glomerular mesangial cells,GMCs)位于肾小球毛细血管袢内,具有类似于血管平滑肌的多种功能,其收缩功能可通过改变毛细血管口径,影响肾小球血流量和滤过面积,调节肾小球滤过率(glomerular filtration rate, GFR)[4]。已有报道,GMCs的功能失调与包括糖尿病慢性肾病在内的多种肾脏疾病相关[5-6]。
钙库操纵的钙内流(store-operated Ca2+entry,SOCE)途径参与引起GMCs的Ca2+内流、产生收缩效应[4,7]。Orai1和STIM1是SOCE的两种主要通道蛋白。我们前期研究证实,STIM1和Orai1表达于GMCs,利用siRNA干扰降低STIM1或Orai1表达可明显抑制原代培养的GMCs的SOCE水平[8]。Ca2+内流,引起局部Ca2+浓度升高,可迅速激活下游信号分子——大电导钙激活的钾通道(large conductance calcium activated potassium channel, BKCa)[9]。研究发现,BKCa也参与调节GFR[10]。但在GMCs中,SOCE与BKCa之间的相互关系以及高糖环境对其的影响并不清楚。本研究拟采用原代培养的大鼠GMCs作为研究对象,揭示GMCs中BKCa-Orai1之间是否存在物理性与功能性的相互作用,以及高糖培养模拟糖尿病对BKCa与Orai1在GMCs表达及其介导的功能的影响,从而为BKCa-Orai1相互作用在DN发病中的作用提供相关的理论依据。
1 材料与方法
1.1材料
1.1.1实验动物 清洁级♂SD大鼠,体质量(150~180) g,由安徽医科大学实验动物中心提供,动物实验严格遵照《实验动物质量管理方法》和《中华人民共和国实验动物管理条例》法规,并经安徽医科大学校动物伦理委员会同意。大鼠分笼饲养于室温(20~24) ℃的环境中,自由摄食与饮水。
1.1.2试剂 毒胡萝卜素(thapsigargin, TG)和Iberiotoxin(IbTX)购于Sigma公司;Fluo-8/AM、Pluronic F-127购于Invitrogen公司;ECL显色试剂盒购于GE healthcare公司;Orai1抗体和BKCa抗体购于Santa Cruz Biotechnology公司;RPMI 1640、胎牛血清购于维森特生物技术有限公司;胰酶购于碧云天生物有限公司。
1.1.3仪器 钙成像荧光显微镜(日本尼康公司);4℃低温离心机(美国Sigma公司);低速离心机(湖南凯达科技仪器有限公司);电泳仪(美国Bio-Rad公司);Bioshine Chemi Q 4600min化学发光成像系统(上海欧翔仪器有限公司)。
1.2方法
1.2.1GMCs原代培养 清洁级♂SD大鼠, 吸入过量CO2麻醉处死,消毒后打开腹腔,迅速取出两侧肾,移至超净台内4℃ PBS中, 去除肾包膜, 将肾皮质部分制成匀浆状, 依次经250、150、120 μm的不锈钢筛网滤过,最后收集120 μm筛网上的颗粒组织,得到肾小球悬液。1 000 r·min-1离心5 min后取沉淀物,加入5 mL含有0.1% Ⅰ型胶原酶的PBS , 震荡水浴箱内37℃孵育30 min , 显微镜下显示肾小球轻度破碎并松动,1 000 r·min-1离心5 min后取沉淀物,加入培养基悬浮,种植于25 cm2的细胞培养瓶中,1 h后更换新鲜培养基,去除杂质,细胞培养液含有RPMI 1640、10%胎牛血清、100 mg·L-1青霉素和100 kU·L-1链霉素。
1.2.2Western blot法检测蛋白表达水平 分别采用正常糖培养基(5 mmol·L-1D-glucose)和高糖培养基(25 mmol·L-1D-glucose)培养GMCs,3 d后移除培养基,加入蛋白裂解液,冰上放置40 min,4℃下12 000 r·min-1离心20 min,留取上清液。以BSA为标准,用Bradford法对上清进行蛋白定量。取20 μg蛋白样品,10% SDS-PAGE电泳,100 V转移1 h至硝酸纤维素薄膜,放入封闭液中37℃封闭1 h;一抗 4℃过夜。β-tubulin作为蛋白加样内参。反复洗膜后,二抗孵育,室温轻摇1 h,洗膜后,用 ECL试剂盒对目的条带进行显影、采集图像,并用Quantity one软件分析测定各带吸光度(A)值作定量分析。
1.2.3免疫共沉淀(co-immunopricipitaiton, co-IP) 原代培养的GMCs加入细胞裂解液,置于冰上40 min,12 000 r·min-1、4℃离心20 min,留取上清液,分别加入3 μg一抗anti-BKCa或anti-Orai1抗体,4℃轻轻震荡2 h,加入Protein agarose A 100 μL,混合后,4℃轻轻震荡过夜。d 2,10 000 r·min-1、4℃离心2 min,弃上清,沉淀用细胞裂解液洗3次。加入100 μL细胞裂解液和50 μL三倍的上样缓冲液。100℃加热样品,离心5 min,上清用于蛋白电泳检测BKCa和Orai1的相互作用。
1.2.4细胞内Ca2+浓度检测 将消化后的GMCs接种于多聚赖氨酸包被后的玻片上,实验前用Fluo-8/AM(终浓度为6 μmol·L-1,且含有0.02% Pluronic F-127)在37℃培养箱中孵育细胞30 min后,用无钙PSS溶液(0 Ca2+-PSS:NaCl 140 mmol·L-1、MgCl21 mmol·L-1、KCl 5 mmol·L-1、EGTA 0.2 mmol·L-1、glucose 10 mmol·L-1、HEPES 4 mmol·L-1,pH 7.4)清洗玻片2遍后,将其安装在钙成像专用浴槽中,并向浴槽中加入500 μL 0 Ca2+-PSS,浴槽置于钙成像荧光显微镜,实时记录胞内游离Ca2+荧光信号。实验过程中,先在0 Ca2+-PSS加入TG(终浓度为 2 μmol·L-1)作用于细胞耗竭钙库,激活钙库操纵的钙内流通道,然后加入Ca2+(终浓度为1 mmol·L-1)引起Ca2+内流,这一过程为SOCE,连续记录20 min。荧光强度反映的是游离钙离子的实时浓度,细胞内Ca2+浓度的变化用荧光相对强度之比(F1/F0)表示。
1.2.5细胞膜电位记录 如上细胞内Ca2+浓度检测实验,GMCs用100 nmol·L-1DiBAC4(3)在室温黑暗处孵育10 min,玻片清洗后安装在专用浴槽中,并向浴槽中加入500 μL 0 Ca2+-PSS,浴槽置于钙成像荧光显微镜,实时记录DiBAC4(3)荧光信号。然后用2 μmol·L-1的TG在0 Ca2+-PSS溶液中处理10 min后,在细胞外液中加入Ca2+(使溶液Ca2+终浓度为2 mmol·L-1),胞内荧光浓度的变化按荧光相对强度之比(F1/F0)进行计算。

2 结果
2.1SOCE引起GMCs膜超极化为检测SOCE对GMCs膜电位的影响,我们利用TG(2 μmol·L-1)耗竭GMCs内钙库,激活钙库操纵的钙通道,10 min后,外液中再加入Ca2+引起外钙内流。结果表明,SOCE可引起GMCs膜电位超极化,且BKCa的阻断剂IbTX(50 nmol·L-1)可以明显抑制SOCE引起的细胞膜超极化水平,说明SOCE引起的钙内流通过激活BKCa引起膜电位超极化(Fig 1)。
2.2Orai1与BKCa在GMCs上的物理性相互作用我们前期实验已证实,Orai1参与介导GMCs的SOCE[8]。为验证SOCE主要通道蛋白Orai1与BKCa在大鼠GMCs上的相互作用,我们将原代培养的大鼠GMCs裂解后,提取蛋白进行免疫共沉淀实验。结果显示,Orai1蛋白与BKCa蛋白之间可以相互沉淀(Fig 2)。结果提示,Orai1与BKCa可能在GMCs上形成了信号复合物,从而使SOCE介导的局部钙浓度的变化可以更加精确、快速地调节细胞膜电位,进而通过负反馈调节控制细胞内的游离Ca2+浓度。
2.3高糖培养抑制SOCE引起的GMCs膜超极化水平为进一步揭示高糖对GMCs的SOCE及其引起的膜电位超极化的影响,我们仍采用TG(2 μmol

Fig 1 Role of BKCa in SOCE-induced membranehyperpolorization of rat GMCs
A: Representative traces showed after treated with 2 μmol·L-1thapsigargin (TG) for 10 min in 0 Ca2+-PSS, membrane hyperpolarization was evoked by 1 mmol·L-1extracellular Ca2+in GMCs with/without 50 nmol·L-1iberiotoxin(IbTX); B: Summary of data showed changes in membrane hyperpolarization in response to extracellular Ca2+(n=4~5).*P<0.05vscontrol.

Fig 2 Co-immunopricipitaion of Orai1 andBKCa in primary cultured rat GMCs (n=3)
Representative images showed coimmunoprecipitation followed by immunoblots. A: Immunoblot with anti-Orai1 antibody; B: Immunoblot with anti-BKCaantibody.
·L-1)耗竭钙库,激活钙库操纵的钙通道,外液中再加入Ca2+引起外钙内流(Fig 1 A)。GMCs经高糖培养3 d后,SOCE引起细胞内Ca2+水平升高增强(Fig 3),而SOCE引起的细胞膜去极化绝对值也增大(Fig 4)。推测这一改变可能与SOCE的主要组成蛋白Orai1和BKCa通道蛋白表达水平的变化有关。

Fig 3 Effect of high glucose(HG) on store-operatedCa2+entry (SOCE) in GMCs
A: Representative SOCE traces was evoked by Ca2+response in GMCs with normal glucose(NG) and high glucose(HG) treatment for 3 days; B: Summary data showed effect of NG and HG in GMCs(n=4~5).*P<0.05vsNG treatment.
2.4高糖培养导致GMCs中Orai1与BKCa表达水平变化为揭示高糖引起的细胞Ca2+水平和膜电位变化的原因,我们进一步检测了高糖培养对SOCE主要通道蛋白Orai1和下游信号分子BKCa的表达水平的影响。结果显示,与正常糖培养相比较,原代培养的GMCs高糖处理3 d后,Orai1和BKCa的表达水平均明显升高(Fig 5)。
3 讨论
血管紧张素Ⅱ、血管升压素、内皮素-1等血管活性物质都可通过SOCE途径,引起GMCs的Ca2+内流、产生收缩效应[4,7]。SOCE通过细胞内钙库的充盈与排空进行调控。当胞内钙库排空,位于内质网膜上的STIM1蛋白N端的钙感受器被激活,发生结构改变并形成多聚体,位于胞质的C端与细胞膜上的Orai1相互作用,激活Orai1通道,引起SOCE[11]。 我们前期研究证实,STIM1和Orai1表达于GMCs,并参与介导SOCE[8]。另外,研究发现,BKCa参与调节GFR[10]。在BKCaβ1亚单位敲除小鼠,基础状态下GFR是正常的,但在血容量增多时,却无法像野生型小鼠一样出现GFR相应增加。从血流动力学方面分析,这种改变可能是因为表达于肾小球入球小动脉[12]和GMCs的BKCa功能异常所致[13]。然而研究表明,在肾小球入球小动脉,BKCa所发挥的抑制血管收缩的作用相对较弱[12],而GMCs的BKCa在对抗其收缩中则发挥主要作用[13]。所以,当敲除BKCaβ1亚基时,BKCa的负反馈调节作用减弱,引起持续的Ca2+内流和GMCs收缩,肾小球滤过面积减小,导致GFR无法根据血容量的增加而增大。
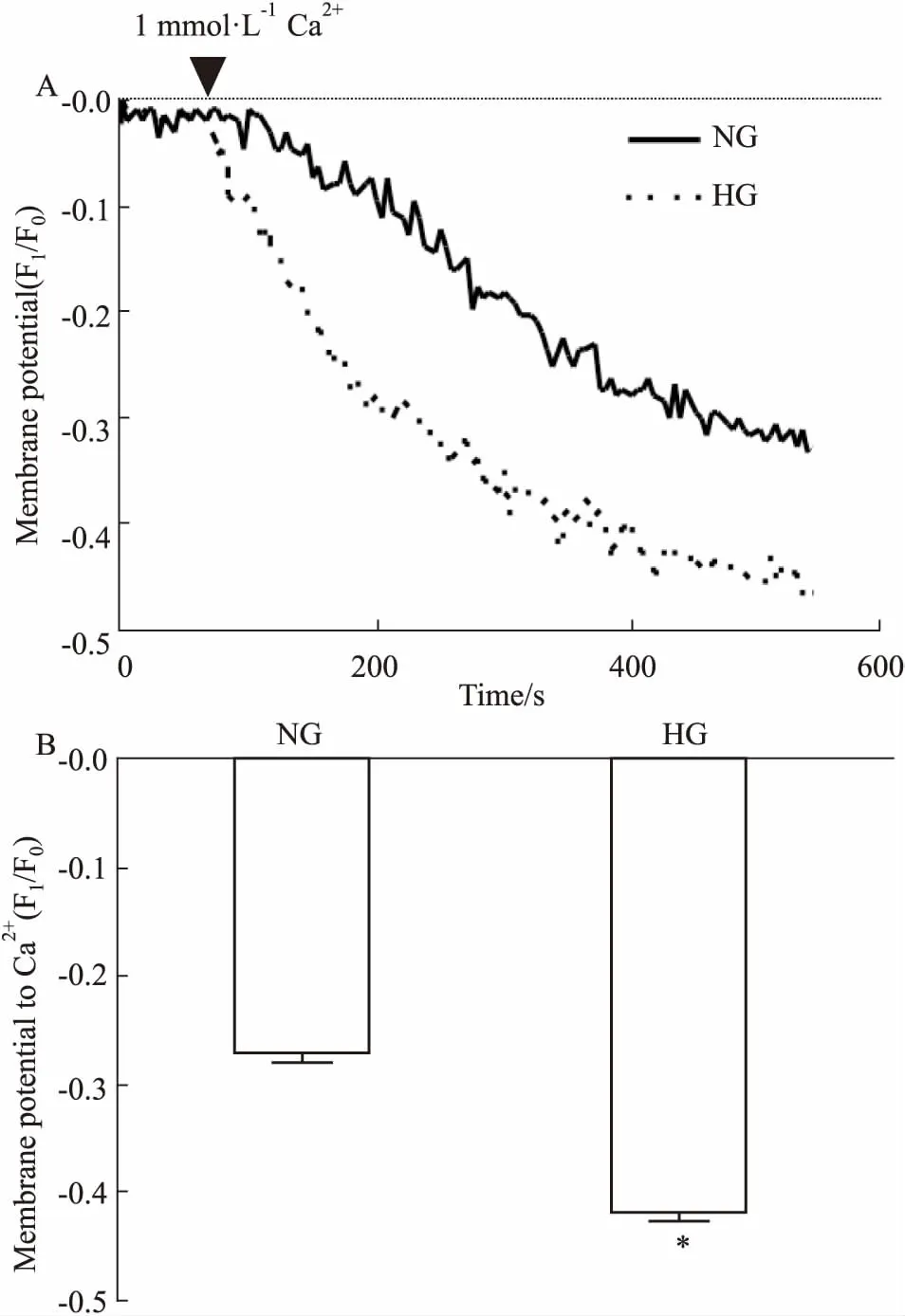
Fig 4 Role of high glucose in SOCE-inducedmembrane hyperpolorization of rat GMCs
A: Representative traces showed after treatment with 2 μmol·L-1TG for 10 min in 0 Ca2+-PSS, membrane hyperpolarization was evoked by 1 mmol·L-1extracellular Ca2+in GMCs with NG or HG; B: Summary of data showed changes in membrane hyperpolarization in response to extracellular Ca2+(n=4~5). *P<0.05vsNG treatment.

Fig 5 Changes of Orai1 and BKCa protein expression induced byhigh glucose in rat primary cultured GMCs n=4)
*P<0.05vsNG treatment
本实验结果显示,在GMCs中,SOCE可以引起细胞超极化,这与Ca2+内流引起膜的去极化的设想不同,说明SOCE激活了其他的通道参与这一过程。进一步的研究发现,BKCa阻断剂IbTX可以明显抑制SOCE引起的细胞膜超极化水平。在细胞信号转导过程中,为了快速、高效地激活下游分子,细胞中参与同一信号转导的几种分子可形成复合物。BKCa既然是SOCE介导的Ca2+内流所激活的下游分子,那么是否可与SOCE通道蛋白形成信号复合物呢?我们利用co-IP检测发现,BKCa可与Orai 1相互沉淀,提示BKCa与Orai 1形成了复合物。根据这些实验结果,推测SOCE介导的钙内流可以通过引起局部Ca2+浓度升高,迅速激活下游信号分子BKCa通道,引起K+外流、细胞膜超极化,形成负反馈而抑制电压依赖性钙通道(voltage-dependence calcium channel, VDCC),导致Ca2+内流减弱,可能参与调节细胞的收缩反应[10]。
糖尿病患者早期肾脏病理改变可能与SOCE功能改变,引起的胞内Ca2+信号异常有关。研究发现,模拟糖尿病微环境的高糖处理可引起GMCs凋亡等改变[14]。最近有研究发现,高糖处理增加SOCE水平和SOCE主要蛋白Orai1、STIM1的表达水平[15]。我们的实验结果发现,在高糖培养3 d后,SOCE的主要通道蛋白Orai1的表达水平和介导的钙内流明显加强,且SOCE引起的GMCs的膜超极化幅度明显增大,继续探究Orai1与BKCa通道蛋白表达水平发现,高糖培养GMCs 3 d增加Orai1与BKCa的表达水平。这些结果提示,高糖培养通过增加Orai1表达水平而使SOCE介导的钙内流增强,并更加高效地激活同样高表达的下游信号分子BKCa通道,引起GMCs膜电位进一步超极化,从而抑制VDCC。这一机制可能参与了肾小球在糖尿病初期功能异常所引起的GFR的改变。但在糖尿病不同时期,GFR的变化有所不同,机制也不尽相同,关于钙信号在糖尿病不同时期对系膜细胞与GFR的影响还有待进一步探讨。
[1] Zac-Varghese S, Winocour P. Managing diabetic kidney disease[J].BrMedBull,2018,125(1):55-6.
[2] Romagnani P, Remuzzi G, Glassock R, et al. Chronic kidney disease[J].NatRevDisPrimers,2017,3:17088.
[3] Moriya T, Tsuchiya A, Okizaki S, et al. Glomerular hyperfiltration and increased glomerular filtration surface are associated with renal function decline in normo- and microalbuminuric type 2 diabetes [J].KidneyInt,2012,81(5):486-93.
[4] Abboud H E. Mesangial cell biology [J].ExpCellRes,2012,318(9): 979-85.
[5] Frische S.Glomerular filtration rate in early diabetes: ongoing discussions of causes and mechanisms [J].JNephrol,2011,24(5): 537-40.
[6] Barton M, Sorokin A. Endothelin and the glomerulus in chronic kidney disease[J].SeminNephrol,2015,35(2):156-67.
[7] Ma R, Du J, Sours S, et al. Store-operated Ca2+channel in renal microcirculation and glomeruli [J].ExpBiolMed,2006,231(2): 145-53.
[8] Shen B, Zhu J, Zhang J, et al. Attenuated mesangial cell proliferation related to store-operated Ca2+entry in aged rat: the role of STIM 1 and Orai 1[J].Age(Dordr), 2013,35(6):2193-202.
[9] Ma R, Pluznick J L, Sansom S C. Ion channels in mesangial cells: function, malfunction, or fiction [J].Physiology, 2005,20: 102-11.
[10] Kudlacek P E, Pluznick J L, Ma R, et al. Role of hbeta1 in activation of human mesangial BK channels by cGMP kinase [J].AmJPhysiolRenalPhysiol, 2003,285(2): F289-94.
[11] Soboloff J, Rothberg B S, Madesh M, et al. STIM proteins: dynamic calcium signal transducers [J].NatRevMolCellBiol,2012,13(9):549-65.
[12] Fallet R W, Bast J P, Fujiwara K, et al. Influence of Ca2+-activated K+channels on rat renal arteriolar responses to depolarizing agonists [J].AmJPhysiolRenalPhysiol,2001,280(4): F583-91.
[13] Stockand J D, Sansom S C. Role of large Ca2+-activated K+channels in regulation of mesangial contraction by nitroprusside and ANP [J].AmJPhysiolCellPhysiol, 1996,270: C1773-9.
[14] 刘 静,张 瑞,李冠青,史永红. SRT1720对高糖诱导的小鼠系膜细胞凋亡的影响[J]. 中国药理学通报,2017,33(8):1164-9.
[14] Liu J, Zhang R, Li G Q, Shi Y H. Influence of STI1720 on apoptosis of high glucose induced mouse mesangial cells[J].ChinPharmacolBull,2017,33(8):1164-9.
[15] Chaudhari S, Wu P, Wang Y, et al.High glucose and diabetes enhanced store-operated Ca2+entry and increased expression of its signaling proteins in mesangial cells[J].AmJPhysiolRenalPhysiol,2014,306(9):F1069-80.
