细菌多元基因编辑技术研究进展及其在合成生物学中的应用
2018-06-07郭明璋刘海燕杜若曦许文涛
郭明璋 刘海燕 杜若曦 许文涛
(1. 中国农业大学北京食品营养与人类健康高精尖创新中心,北京 100083;2.华北理工大学公共卫生学院,唐山 063210)
许多技术的发展都有一个从低通量到高通量的过程。高通量技术通常具有全面、准确、高效和低廉等优势。在信息科学领域,大数据技术的发展实现了对信息的高通量分析,已经改变了人类生活的各个方面[1];在生物技术领域,高通量测序技术的出现及不断创新发展从根本上改变了基因组学、转录组学、宏基因组学甚至整个生物学领域的科学研究思路和方式[2-4]。基因编辑技术是继基因测序技术后又一项具有广泛应用前景的热点生物技术。科学家在通过基因测序技术对生物的基因组进行“阅读”后,必然会希望通过基因编辑技术对生物基因组进行“改写”[5],以研究基因的功能与特性,进而对基因进行改造和优化。然而与基因测序的高通量程度相比,基因编辑的高通量化尚处在探索和雏形阶段(图1)。基因编辑高通量化的实现将使基因编辑技术实现质的飞跃,也将是推进基因编辑产业化的重要动力。

图1 基因测序技术和基因编辑技术发展进程
多元基因编辑技术是实现高通量基因编辑的重要中间过程,类似于测序技术的发展历程中的利用毛细管电泳进行多重平行测序阶段(如ABI公司3730XL型号的96道毛细管测序仪)。多重平行测序是利用传统Sanger测序的原理,通过测序设备的改进与优化,实现了测序效率的提升[6]。同样,目前最为成熟的微生物多元基因编辑技术——基因组多重位点自动改造技术(Multiplex automated genome engineering,MAGE)[7],也是利用传统单一基因编辑技术的原理,通过自动化循环设备的设计,实现了基因编辑效率的提升。无论从基因编辑的靶基因数量级,还是从基因编辑原理的突破性来看,目前的多元基因编辑技术尚不能称作“高通量”。从“单一”到“多元”是量变的过程,而从“多元”到“高通量”是质变的过程。现阶段的多元基因编辑正在为高通量基因编辑的出现积累着技术创新。
与基因测序技术不同,基因编辑技术在不同生物中的发展存在很大差异。同动植物相比,细菌的基因编辑极有可能更早实现多元和高通量。首先,随着细菌合成生物学的迅速发展,研究者经常需要对细菌基因组进行全方位编辑,甚至改变细菌基因组的密码子系统[8],这使高通量基因编辑技术在细菌领域具有更加迫切的需求,迫切的需求往往推动着新技术的发展。其次,细菌易于培养,繁殖迅速,可以缩短研究时间,且细菌群落中个体数量庞大,更易筛选到目标个体。再次,相较于动植物基因编辑,细菌基因编辑的方法更加多样,实现高通量基因编辑的选择性更多。
本文综述了近10年来细菌领域多元基因编辑技术的发展,对基于各种原理的多元基因编辑技术进行比较,并探讨了它们进一步实现高通量基因编辑的可能性。在此基础上,本文对多元基因编辑技术在微生物合成生物学中的应用进行了介绍和展望。
1 细菌基因编辑的基本工具与原理
1.1 位点特异性基因重组系统
Cre-lox重组系统和FLP-FRT重组系统都是位点特异性基因重组技术。Cre-lox系统来源于大肠杆菌(Escherichia coli)P1噬菌体,可以促使噬菌体DNA插入到细菌基因组中[9-10]。Cre为重组酶,可以识别长度为34 bp具有方向性的loxP位点。同一DNA分子上同向的两个loxP位点发生重组,可以删除两个loxP位点之间的DNA片段;同一DNA分子上反向的两个loxP位点发生重组,可以使两个loxP位点之间的DNA片段发生倒置;具有单个loxP位点的环状DNA序列与第二个DNA分子的loxP位点发生重组,可以将环状序列插入到第二个DNA分子的loxP位点上。loxP位点在自然界基因组中出现的概率极低,因此在进行基因编辑时,首先要在受体基因组上引入loxP位点,再诱导Cre重组酶的表达并催化loxP位点之间发生重组。在细菌基因编辑中,Cre-lox重组系统常用作删除选择标记基因、大片段基因的删除和倒置等。Cre重组酶发挥作用不需要任何辅助因子,Cre-lox重组系统具有广谱性,理论上可以在任何细胞环境的任何类型DNA分子上发挥有效作用。Flp-FRT系统是Cre-lox系统在真核细胞内的同源系统,与Cre-lox系统的工作原理极为相似,Flp为重组酶,识别具有方向性的FRT位点(图2)[11]。
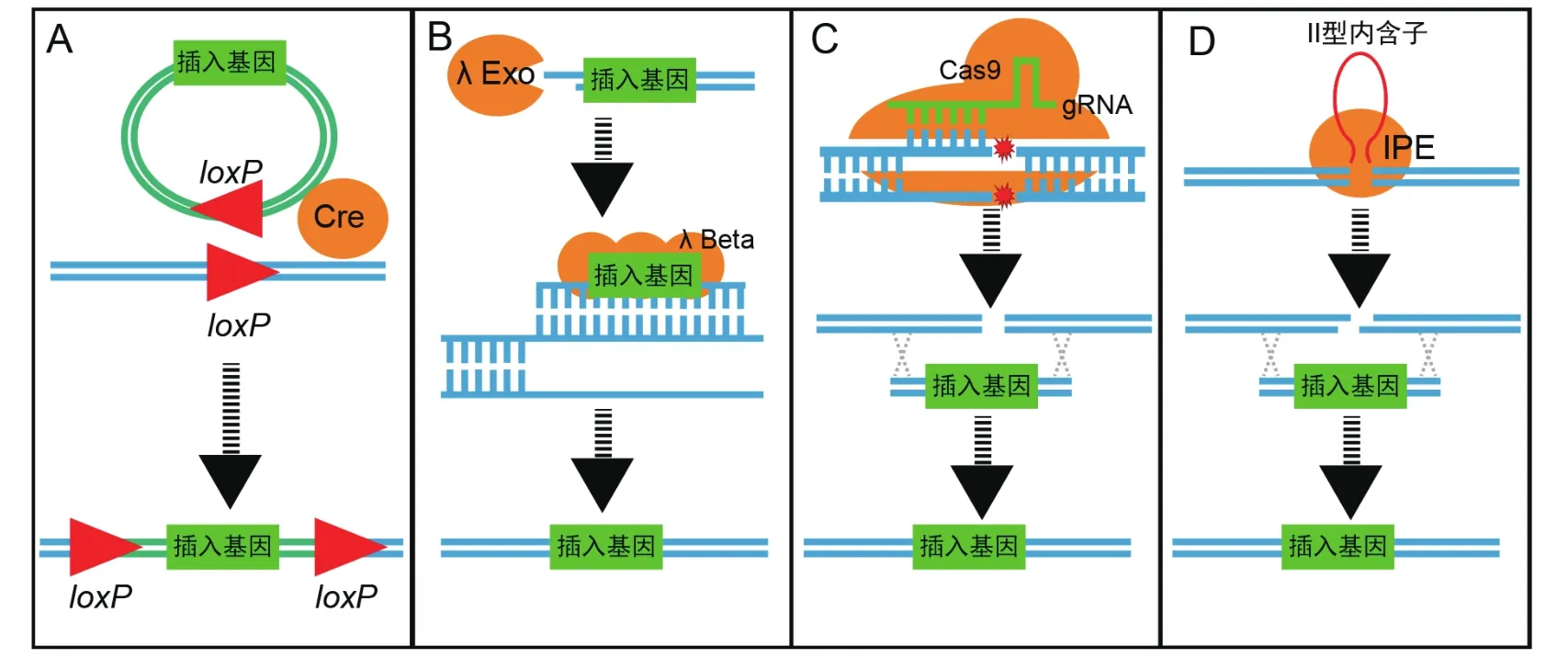
图2 基因编辑基本工具的工作原理
1.2 同源重组系统
λ-Red同源重组系统包含3个来源于λ噬菌体Red操纵子的基因,其中λ Gam蛋白抑制RecBCD酶的活性,以保护外源线性DNA免受降解。λ Exo是以双链DNA(Double-stranded DNA,dsDNA)为底物的核酸外切酶,可以催化5'-3'方向的降解,而产生3'-5'的线性单链DNA(single-stranded DNA,ssDNA),当dsDNA的一条链被降解后,产生的ssDNA将不会被λ Exo降解。λ Beta蛋白可以结合在ssDNA上起到保护作用,引导同源配对促进同源重组的发生[12]。RecE/T重组系统与λ-Red同源重组系统作用机制类似,RecE蛋白发挥与λ Exo蛋白相似的功能,RecT蛋白发挥与λ Beta蛋白相似的功能[13]。RecE/T系统缺少RecBCD酶的抑制蛋白。进行基因编辑时,在细菌中引入两端具有与受体基因组DNA同源片段的外源dsDNA分子,借助于λ-Red重组系统或RecE/T重组系统,诱发同源片段中间的外源DNA序列与基因组序列发生交换,从而可实现基于同源重组的基因编辑。
传统的λ-Red同源重组系统是在细菌中引入dsDNA,Ellis等[14]在2001年提出利用70-90 bp长度的ssDNA为底物可以简化λ-Red系统,不需要λ Exo介导的产生单链的过程。目前比较接受的机制为退火学说(Strand annealing),即无论是在细菌中引入同源dsDNA后通过λ Exo外切酶产生的ssDNA,还是在细菌中直接引入的ssDNA,都会在λ Beta蛋白保护和介导下以冈崎片段的方式结合到DNA复制过程中的后随链上,产生一个野生型基因组和一个异源双链的基因组。随后具有异源双链基因组的细菌细胞再经过一次DNA复制和细胞分裂产生一个野生型基因组和一个突变型基因组[15]。
1.3 靶向核酸酶
锌指核酸酶(Zinc finger nuclease,ZFN),转录激活样效应物核酸酶(Transcription activator-like effectors nuclease,TALEN)和成簇的规律间隔的短回文重复序列(Clustered regularly interspaced short palindromic repeats,CRISPR)/Cas9核酸酶是在基因编辑中应用最为广泛的3种核酸酶。ZFN通过锌指结构识别特定的核酸序列,通过Fok I核酸内切酶活性切割DNA[16];TALEN通过转录激活样效应物识别特定的核酸序列,通过Fok I核酸内切酶活性切割DNA[17];目前的CRISPR/Cas9通过向导RNA(guide RNA,gRNA)识别特定的核酸序列,通过Cas9的核酸酶活性切割DNA[18]。靶向核酸酶在特定位点对DNA进行切割,产生了DNA双链断裂(Doublestrand DNA break,DSB),在原核细胞中DSB的产生可促进该位点同源重组的发生率,在真核细胞中还可通过非同源末端连接修复产生移码突变而实现基因的功能性敲除。此外,在真核细胞中通过改造的Cas9与碱基编辑酶进行蛋白融合,还可实现对特定区域的碱基进行定点编辑。
1.4 可移动II型内含子
可移动II型内含子(Mobile group II introns)广泛存在于细菌和真核生物细胞中,是位点特异性逆转录因子。II型内含子具有核酶活性,经过自剪接后脱离mRNA,形成套索结构,并可以编码内含子编码蛋白(Intron-encoded protein,IEP)。IEP可以结合在内含子上形成核糖核蛋白复合物并入侵特定的DNA位点,通过核酶位点的反向剪接能力将自身整合到DNA一条链上,随后通过IEP的反转录活性形成完整的DNA双链,这一过程被称为“归巢(Retrohoming)”。II型内含子通过其EBS1和EBS2区域的碱基与靶标DNA的进行互补配对而实现对特定靶标DNA位点的识别,因此可以通过编辑EBS1和EBS2区域的碱基序列,改变II型内含子识别位点[19]。在细菌中,通过在编码基因中插入II型内含子,可以实现对该基因的功能性敲除[20]。
2 细菌多元基因编辑的基本策略
与单个基因编辑相比,多元基因编辑的成功率较低。从概率上分析,若每一个基因编辑的成功率为pi,则使n个相互独立的基因在同一个细胞内全部编辑成功率为PM=。多元编辑当n很大时,PM值将非常小。若想要提高PM值,理论上有两种基本策略:一是提高每个基因编辑的效率使pi更接近于1;二是减少n的值(图3)。
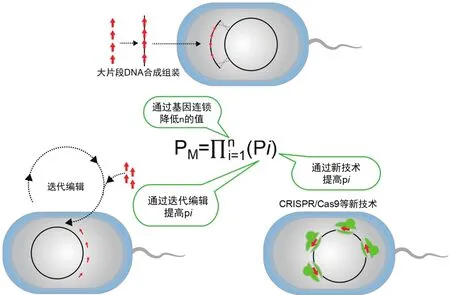
图3 提高细菌多元基因编辑效率的基本策略
提高pi可以通过多种方式实现。首先,最根本的方式是改进基因编辑技术,包括对原有基因编辑技术的参数优化,和开发新的高效基因编辑技术。其次,在已有基因编辑技术不提升的情况下,pi值的提高可以通过迭代来实现。在同一个细胞及其子代中某一位点进行N次迭代基因编辑,则该位点基因编辑成功的概率为p’=1-(1-p)N。由此可见,迭代次数N越大,则p’越接近于1,多元基因编辑成功率PM越大。此外,在无法改变基因编辑效率的情况下,可以通过选择压力杀死未成功编辑细胞,以提高成功编辑的细胞的比例。在多元编辑中,不可能针对每一个基因靶标设计一个选择压力,否则一方面会在基因组上留下过多的选择标记;另一方面当基因编辑效率较低时,多种选择压力可能会杀死所有细胞。但由于“共选择”和“共转化”等现象的存在,可以通过适当的设计,通过少量的选择标记完成对所有基因编辑的选择。
降低n的数值可以通过将多个靶标基因进行连锁来实现。对于基因敲入,可以将多个基因整合到一个操纵子上。对于受体基因组上不连续位点的基因敲除或者基因编辑,则可以通过基因合成的方法构建跨越多个靶基因位点的同源大片段,然后将合成的大片段与受体基因组进行同源重组。
3 基于迭代编辑的方法
3.1 MAGE技术和CAGE技术
多元自动基因编辑(MAGE)技术是由Wang和Isaacs等[7]于2009年提出的。MAGE的基本思想是通过迭代提高多元基因编辑的成功率。迭代策略在理论上适用于任何基因编辑技术,但迭代基因编辑也会使工作量和工作时间成倍增加。他们选择了操作相对简单的λRED系统,并以人工合成的ssDNA为同源重组反应底物。MAGE技术需要在受体大肠杆菌的基因组上整合温控启动子控制表达的λRED系统(Exo、Beta和Gam),并敲除基因组上mutS基因以抑制细菌错配修复。MAGE的每次基因编辑操作包括以下步骤:(1)细胞培养至对数中期;(2)在42℃诱导λ Beta蛋白的表达;(3)将细胞置于4℃;(4)用无菌蒸馏水洗去培养基并重悬细胞;(5)加入ssDNA;(6)电转化;(7)恢复培养,重新回到步骤1进入下一个循环[21]。MAGE可以通过设备实现自动化循环。MAGE的效率可以高达每个循环完成35%的基因编辑,随着编辑区域(不匹配区域)的增长,编辑效率会有所下降。当进行每个靶位点18个碱基的插入时,MAGE的每个循环的编辑效率为 2%-5%[22]。
2011年,Isaacs等[23]在对大肠杆菌全基因组314个终止密码子TAG进行编辑时,又提出(Hierarchical conjugative assembly genome engineering,CAGE)的策略,即将整个基因组DNA划分为32个区域,其中31个区域包含10个终止子靶标,另一个区域包含4个终止子靶标。进行基因编辑时,先在不同细胞群中对每个区域内的TAG终止子分别进行编辑,再将编辑好的基因组区域通过接合方式进行分层合并,最终产生基因组中同时包含32个成功编辑区域的细胞。作者设计CAGE,而非在一次MAGE中加入所有314个TAG终止子编辑同源ssDNA的目的,主要是通过减少每个MAGE的靶标数。每次MAGE反应的靶标数减少,不仅可以减少λRED系统工作时导致的细菌基因组非期望突变的增加,而且便于对靶标的筛选和重组率的计算。CAGE技术可以实现微生物全基因组的精准编辑[24]。
3.2 CoS-MAGE技术
共选择多元自动基因编辑(CoSelection MAGE,CoS-MAGE)是对MAGE的改进。在对MAGE研究的过程中,研究者们无意发现在每一次MAGE循环过程中,都有某些菌落的细菌中包含不止一个位点的基因编辑,这暗示了当细胞中发现某一个突变位点,则该细胞中发现其他突变位点的概率会增加[25],该现象有可能缘于某些细胞亚群可能更适于电转化或更适于ssDNA的同源重组,这一现象被称为CoSMAGE[25-26]。因此,可以在作为靶标基因同源重组底物的ssDNA寡核酸库中加入小部分共选择寡链核苷酸(CoS oligo),将基因组上失活的选择标记(galK-,malK-,cat-和bla-)进行回复突变,以通过营养选择或抗生素选择及共选择现象,提高MAGE中成功编辑细胞的比例。对12个靶基因编辑实验结果表明,经过2个MAGE循环的基因编辑,传统MAGE的阳性率为2%-3%,而CoS-MAGE的阳性率可以达到25%。进一步研究表明,当CoS oligo的作用位点和靶标基因位点之间的距离在500 kb之内时,可以在一个循环内通过1个CoS oligo的筛选作用获得8个靶标基因的编辑体。
3.3 pORTMAGE
由于MAGE依赖于ssDNA结合到复制的基因组上,形成异源双链,因此在MAGE过程中,必须要抑制细菌内源错配修复机制。传统的MAGE中使用mutS基因突变的菌株,因此限制了MAGE使用的广谱性,并且提高了背景基因组发生非期望突变的概率。2016年Nyerges等[27]开发了pORTMAGE质粒,该质粒可以温控表达催化位点失活的MutL基因。MutL蛋白是MutHLS复合物的组分之一,催化位点失活的MutL蛋白可以破坏MutHLS的功能。当催化位点失活的MutL基因不表达时,野生型MutL蛋白可以使细菌恢复错配修复机制,而降低背景基因组的非期望突变率。因此,pORTMAGE可以使野生型细菌菌株实现低背景错配率的MAGE循环。
3.4 MuGENT
MAGE技术及其衍生出的CoS-MAGE技术和pORTMAGE技术都是将迭代编辑思想应用在ssDNA介导的基因重组,这一过程需要λRED系统的催化和细菌错配修复机制的抑制。Dalia等[28]于2014年提出了将迭代编辑的思想与转化(Transformation)相结合的MuGENT技术方案。转化在很多细菌中都会自然发生,与ssDNA介导的基因重组相比,基于转化的基因编辑对受体细胞没有基因型的要求,应用的菌株范围更加广泛。与CoS MAGE的共选择效应类似,转化的过程中也会发生非连续基因的共转化。MuGENT可基于共转化现象通过标记基因的筛选而提高获得靶基因成功编辑菌群的概率。Dalia等将MuGENT应用在革兰氏阴性的霍乱弧菌(Vibrio cholerae)和革兰氏阳性的肺炎链球菌(Streptococcus pneumoniae)上,都取得了较高的成功率。
3.5 TRMR-MAGE
可追溯多元编辑(Trackable multiplex recombineering,TRMR)是Warner等[29]在2010年提出的用于研究基因功能的方法。该方法并非实现在一个细胞中进行多基因编辑,而是针对细胞群中的每一个细胞进行不同靶标的单基因编辑,提升靶标基因的表达量,之后将细胞群置于选择压力下,对选择压力有抗性的细胞亚群将会得到富集,而它们基因组中标记的靶标基因也因而得以富集。通过对靶标基因比例变化分析,可以分析与此选择压力相关的功能基因。
Sandoval等[30]在 2012年提出 TRMR 可以与MAGE联用。严格来讲,TRMR-MAGE并不是多元编辑技术的创新。与其他MAGE的衍生技术不同,TRMR-MAGE并非优化MAGE技术的成功率,而是通过TRMR来筛选MAGE的靶标。利用TRMR分析与某一选择压力相关的基因,再通过MAGE技术同时编辑这些基因,从而获得对该选择压力的高抗性菌株。
4 应用CRISPR/Cas9核酸酶的多元基因编辑方法
提高单个基因编辑的成功率pi的方法最根本的是改进基因编辑工具。CRISPR/Cas9核酸酶系统来源于细菌的免疫防御系统,其高效性和在真核生物中的广泛应用激发了微生物学家的兴趣。CRISPR/Cas9在细菌单个基因编辑中有许多成功的案例,如Jiang等[31]利用CRISPR/Cas9系统编辑了大肠杆菌中的rpsL基因,成功率达到65%。CRISPR/Cas9核酸酶系统对识别位点切割产生的DSB对于许多细菌来说往往是致命的[32],在某些细菌中,单个基因的DSB可以通过同源重组或非同源末端连接的方式进行修复并促进基因编辑的发生,但对于多元基因编辑来说,CRISPR/Cas9系统产生的多个DSB通常会导致细菌死亡。因此,目前将CRISPR/Cas9系统应用在多元基因编辑的案例并不多。
4.1 CRMAGE技术
2015年,Ronda等[33]提出了在 MAGE技术中应用CRISPR/Cas9靶向野生型基因,切割未发生同源重组的基因组,使野生型细菌死亡,从而提高MAGE中基因编辑细菌的比例,这种CRISPR/Cas9与MAGE联用的策略被命名为CRMAGE。对于3个靶标基因的编辑,CRMAGE可以将成功率从MAGE技术的0.68%-5.4%提升到96.5%-99.7%。
4.2 CRISPRNickases
野生型Cas9蛋白有两个活性位点,分别切割DNA的两条链,从而产生DSB。如果利用野生型Cas9蛋白和靶向多个位点的sgRNA进行多元基因编辑,会在基因组上产生多个DSB。大部分细菌无法及时对多个DSB进行修复而导致基因组片段化,致使细胞死亡(图4)。当将Cas9的一个切割位点突变失活后(如Cas9D10A),Cas9将只能切割DNA的一条链而变成切刻酶[34]。如图4所示,切刻版Cas9产生单链DNA断裂(Single Stranded DNA Breaks,SSB),由于另一条链的连接作用,即使在基因组上产生多个SSB,也不会导致基因组片段化,因此对细菌来说是非致命的。基因组上SSB的产生可以促进同源重组的发生,因此切刻版Cas9可以应用在多元基因编辑中[32]。Standage-Beier[32]于 2015 年报道了切刻版Cas9用于大肠杆菌多位点基因敲除的研究,该研究将多个sgRNA整合到质粒pSG4K5上,通过对多位点的切刻,促进了切刻位点附近的同源重组的发生,删除了133 kb的基因组DNA片段。
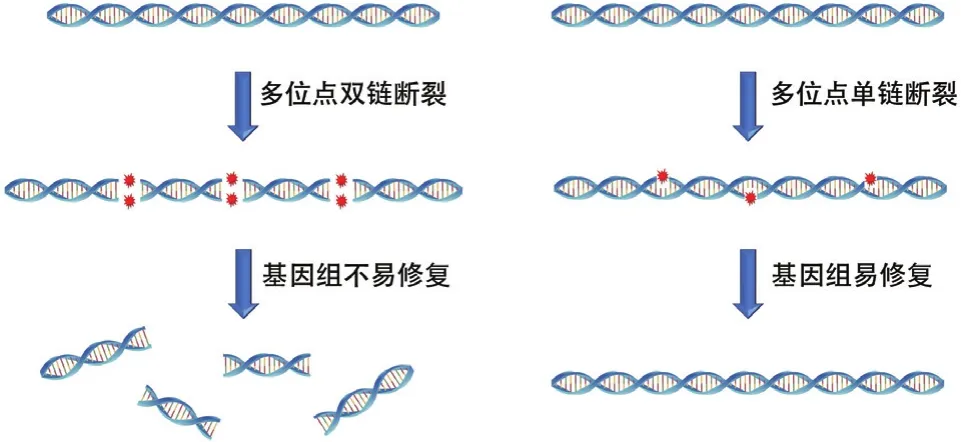
图4 多位点双链断裂与多位点单链断裂比较示意图
4.3 Orthogonal Cas9技术
除了野生型Cas9作为核酸酶及改造Cas9作为切刻酶外,还可以对Cas9进行其他形式的改造以形成Cas9激活子(Cas9 Activator)来激活基因表达、或Cas9抑制子(Cas9 Inhabitior)来抑制基因表达等。但是在同一细胞内,源于同一种Cas9蛋白的不同改造型不能同时发挥作用。例如,一个细胞基因组中整合了来自于化脓性链球菌(Streptococcus pyogenes)的Cas9激活子后,无法用S. pyogenes野生型Cas9进行基因编辑。Esvelt等[35]研究了来自不同的细菌[(S. pyogenes),嗜热链球菌(Streptococcus thermophilus),脑膜炎奈瑟氏菌(Neisseria meningitidis)和齿垢密螺旋体(Treponema denticola)]Cas9的对识别位点序列的偏好性,开发了Orthogonal Cas9方法,利用不同细菌来源的Cas9蛋白,同时对一个细胞的不同基因进行基因编辑和基因调控。
5 基于大片段DNA合成组装的多元基因编辑
Krishnakumar等[36]于2014年报道了通过大片段DNA合成而实现多元编辑的案例。他们首先人工合成了长达72 kb的dsDNA片段,通过Cre-lox重组系统将此人工合成的片段与大肠杆菌基因组上126 kb的同源片段进行重组,重组后实现了对126 kb内多个不连续基因的敲除,最后再通过λRED系统去除基因组上的lox位点,从而实现无痕操作。此过程相当于把多个小片段基因编辑操作转变为一个大片段基因编辑操作,人为将基因组上多个不连续基因进行连锁,从概率的角度看,也相当于破坏了各个基因敲除成功事件之间的独立性。
与迭代方法相比,基于大片段DNA合成的多元基因编辑耗时更短,适用细菌范围更广,并且可以实现基因敲除、基因敲入、碱基替换等多种形式的基因编辑。随着DNA合成费用的降低,这一方法将有更广泛的应用。
6 细菌多元基因编辑技术在合成生物学中的应用方向
6.1 细菌基因组无害化和最小化
现阶段合成生物学多数是利用细菌基因组作为功能系统构建的骨架。细菌基因组一般包含2-6 Mb个碱基,虽然比真核生物基因组小的多,但细菌基因组中包含许多毒力基因使细菌具有病原性,包含许多重组DNA和可移动DNA使细菌基因组具有不稳定性,包含许多无意基因、非必需基因和未知功能基因增加了基因组的复杂性,并且可能会占用一定的核苷酸、氨基酸、核糖体等资源,影响合成生物学系统的工作。因此,自然界中的细菌菌株或只经过少量突变改造的细菌菌株并非合成生物学的理想受体。将细菌基因组中非必需基因进行删除,得到无害化和最小化的细菌基因组,作为合成生物学基因编辑的受体骨架,将有利于获得安全、稳定、高效的合成生物学系统。
Pósfai等[37]于2006年报道了对大肠杆菌K-12菌株基因组的最小化处理,通过删除非必需基因、重组基因和可移动原件和潜在的毒理基因,使基因组减小了15%的长度。研究者意外的发现,基因组可以使菌株产生高电转效率和重组的准确率。此研究并未采用多元基因编辑的方法,而是对每一个靶标片段分别进行敲除,工作量较大。现阶段,通过CRISPRNickases技术的多元靶标同时敲除,可以高效的完成基因组最小化。
6.2 细菌基因组中大片段插入
合成生物学需要在受体细胞中引入新的基因元件以使细胞完成相应的功能。目前大部分合成生物学案例将新基因元件置于高拷贝质粒上,而非整合到受体基因组中,这样设计的优点是操作简便,功能基因元件的表达量高,但也存在功能元件表达量不稳定,易丢失等缺点,并且质粒具有更高的细胞间转移的风险,存在环境安全性隐患。因此将功能元件整合到基因组上可能是未来合成生物学发展趋势。外源多元基因插入可以通过大片段DNA合成整合技术实现。
6.3 细菌基因组密码子系统改造
大部分生物的基因组都采用同一套密码子系统,因此通常情况下外源基因可以在受体细胞中正常表达。当某些外源基因表达的蛋白质中含有稀有氨基酸时,受体细胞中没有与稀有氨基酸相对应的密码子和tRNA,则需要对受体基因组的密码子进行改造。例如,产甲烷古菌的甲基转移酶可以用于生物能源的生产,该酶中含有稀有氨基酸——吡咯赖氨酸,在古菌中由UAG编码[38]。UAG在细菌中是终止密码子,因此若要在细菌中表达甲基转移酶以生产甲烷,则需要将细菌受体基因组中原有的UAG全部改为UAA或UGA,敲除受体基因组上的释放因子1(Release Factor 1),并引入吡咯赖氨酸转运系统。Isaacs等[23]通过MAGE-CAGE技术,成功的将大肠杆菌菌株基因组中所有的314个TAG终止密码子改写为TAA。
通用的密码子系统使用64个密码子编码20个氨基酸,其中存在密码子简并现象。密码子简并可以减少有害突变,维持物种稳定性,但也导致了密码子信息的浪费。Ostrov等[39]则通过大片段合成与λRED系统相结合的方法,构建了基因组中只使用57个密码子的大肠杆菌。7个未使用的密码子AGA、AGG、AGC、AGU、UUA、UUG 和 UAG 便可以用于转运稀有氨基酸或其他特殊氨基酸。随着人类对蛋白结构认识的不断深入,在蛋白质中引入特殊氨基酸可能会使蛋白质功能更加丰富。
6.4 受体细菌基因组代谢物的调控
相比于人工配制的缓冲液体系,微生物细胞质液是十分复杂的环境,其中的代谢物种类和浓度会影响合成生物学菌株的功能效率。在合成生物学设计时,可能需要对宿主某些代谢物的产量进行调控。例如细菌细胞中某些必需代谢物会抑制功能元件的活性,不能通过基因敲除完全去除,只能通过调低这些抑制物生产相关的基因,降低抑制物在细胞质中的浓度。又如细胞中的某些代谢物可能有助于功能元件的活性,需要调高相关基因的表达,以提高功能元件的效率。生物体内代谢物的产量通常受多个基因的调控,调节这些基因的表达量,可以通过编辑它们的启动子实现。Wang等[25]利用CoSMAGE技术,将12个与芳香族氨基酸衍生物合成相关基因的启动子同时编辑为T7启动子,成功优化了芳香族氨基酸衍生物的产量。
7 总结与展望
高通量基因编辑技术具有广泛的应用前景,与合成生物学相结合,可能再一次改变人类的生活与健康[40-41]。目前三种类型的多元基因编辑技术虽然已取得许多重要的进展,但都存在各自的缺陷,影响其进一步高通量化的发展。迭代的方法可以实现多元基因敲除、基因编辑和短片段的基因敲入,但由于需要多轮的基因编辑,因此耗费时间较长,且受体基因组容易产生背景突变。开发和应用新基因编辑工具是推动高效的多元基因编辑技术发展的根本动力,目前的CRISPR-Cas9技术虽然催化基因编辑的效率很高,可以实现对微生物的高效基因敲除与基因编辑,但由于其对部分微生物的致死性,难以成为广谱性的微生物多元基因编辑技术,且CRISPR-Cas9技术并不能做到对任意位置的基因组进行编辑。基于大片段DNA合成组装的多元基因编辑可以实现多元基因敲入。目前受限于DNA合成的准确性及对基因组结构认识的不完整,几个碱基的合成错误就可能导致细胞无法正常生存,因此现阶段的技术还无法做到全基因组的合成,只能合成部分大片段然后组装到基因组上。
若想要进一步实现高通量基因编辑,则需要在两方面取得突破性进展。一方面要有新的高效的基因编辑工具出现,这种新技术需要能做到90%以上的基因编辑效率,不影响宿主细胞的基因修复系统,且可以实现多种类型的基因编辑。另一方面,高效的基因编辑工具通常以外源核酸片段指导靶向性,因此需要开发高效的多元核酸片段连锁转化方法。例如将所有的核酸片段包裹在囊泡中,然后与去壁的细菌细胞进行融合;或者将核酸片段整合到一个质粒上,然后用CRISPR/Cas9等核酸酶切割产生线性片段。随着DNA合成技术的不断发展,以及人类对生物基因组结构认识的不断深入,全基因组合成也可能会逐步成为合成生物学中常用的技术。如果将高通量基因编辑技术称为第二代基因编辑技术,类比基因测序技术的发展历程,全基因组合成可能称为第三代基因编辑技术。
[1]任向楠, 丁钢强, 彭茂祥, 等. 大数据与营养健康研究[J]. 营养学报, 2017, 39(1):5-9.
[2]杜玲, 刘刚, 陆健, 等. 高通量测序技术的发展及其在生命科学中的应用. [J]. 中国畜牧兽医, 2014, 2014((12)):109-116.
[3]郭明璋, 许文涛. 基于高通量测序技术的细菌非编码 研究方法进展[J]. 生物技术通报, 2015, 31(4):99-104.
[4]李东萍, 郭明璋, 许文涛. 测序技术在肠道微生物中的应用研究进展[J]. 生物技术通报, 2015, 31(2):71-77.
[5]Boeke JD, Church G, Hessel A, et al. The genome projectwrite[J]. Science, 2016, 353(6295):126-127.
[6]Swerdlow H, Wu S, Harke H, et al. Capillary gel electrophoresis for DNA sequencing:laser-induced fluorescence detection with the sheath flow cuvette[J]. Journal of Chromatography A, 1990, 516(1):61-67.
[7]Wang HH, Isaacs FJ, Carr PA, et al. Programming cells by multiplex genome engineering and accelerated evolution[J]. Nature, 2009,460(7257):894-898.
[8]Amiram M, Haimovich AD, Fan C, et al. Evolution of translation machinery in recoded bacteria enables multi-site incorporation of nonstandard amino acids[J]. Nat Biotechnol, 2015, 33(12):1272-1279.
[9]Sternberg N, Hamilton D. Bacteriophage P1 site-specific recombination:IRecombination between loxP sites[J]. Journal of Molecular Biology, 1981, 150(4):467-486.
[10]Sternberg N. Bacteriophage P1 site-specific recombination:IIIStrand exchange during recombination at lox sites[J]. Journal of Molecular Biology, 1981, 150(4):603-608.
[11]Cox MM. The FLP protein of the yeast 2-microns plasmid:expression of a eukaryotic genetic recombination system inEscherichia coli[J]. Proc Natl Acad Sci USA, 1983, 80(14):4223-4227.
[12]Murphy KC. Use of bacteriophage λ recombination functions to promote gene replacement inEscherichia coli[J]. J Bacteriol,1998, 180(8):2063-2071.
[13]Zhang Y, Buchholz F, Muyrers JPP, et al. A new logic for DNA engineering using recombination inEscherichia coli[J]. Nature Genetics, 1998, 20(2):123-128.
[14]Ellis HM, Yu D, DiTizio T. High efficiency mutagenesis, repair,and engineering of chromosomal DNA using single-stranded oligonucleotides[J]. Proc Natl Acad Sci USA, 2001, 98(12):6742-6746.
[15]Pines G, Freed EF, Winkler JD, et al. Bacterial recombineering:genome engineering via phage-based homologous recombination[J]. ACS Synthetic Biology, 2015, 4(11):1176-1185.
[16]Beumer K, Bhattacharyya G, Bibikova M, et al. Efficient gene targeting in Drosophila with zinc-finger nucleases[J]. Genetics,2006, 172(4):2391-2403
[17]Zu Y, Tong X, Wang Z, et al. TALEN-mediated precise genome modification by homologous recombination in zebrafish[J].Nature Methods, 2013, 10(4):329-331.
[18]Jakočiūnas T, Jensen MK, Keasling JD. CRISPR/Cas9 advances engineering of microbial cell factories[J]. Metabolic Engineering,2016, 2016((34)):44-59.
[19]Lambowitz AM, Zimmerly S. Group II introns:mobile ribozymes that invade DNA[J]. Cold Spring Harbor Perspectives in Biology, 2011, 3(8):a003616-a003616.
[20]Chen Y, McClane BA, Fisher DJ, et al. Construction of an alpha toxin gene knockout mutant of Clostridium perfringens type A by use of a mobile group II intron[J]. Applied and Environmental Microbiology, 2005, 71(11):7542-7547.
[21]Gallagher RR, Li Z, Lewis A O, et al. Rapid editing and evolution of bacterial genomes using libraries of synthetic DNA[J]. Nature Protocols, 2014, 9(10):2301-2316.
[22]Wang HH, Huang PY, Xu G, et al. Multiplexedin vivoHis-tagging of enzyme pathways for in vitro single-pot multienzyme catalysis[J]. ACS Synthetic Biology, 2012, 1(2):43-52.
[23]Isaacs FJ, Carr PA, Wang HH, et al. Precise manipulation of chromosomesin vivoenables genome-wide codon replacement[J]. Science, 2011, 333(6040):348-353.
[24]Ma NJ, Moonan DW, Isaacs FJ. Precise manipulation of bacterial chromosomes by conjugative assembly genome engineering[J].Nature Protocols, 2014, 9(10):2285-2300.
[25]Wang HH, Kim H, Cong L, et al. Genome-scale promoter engineering by coselection MAGE[J]. Nature Methods, 2012, 9(6):591-593.
[26]Carr PA, Wang HH, Sterling B, et al. Enhanced multiplex genome engineering through co-operative oligonucleotide co-selection[J].Nucleic Acids Research, 2012, 40(17):e132-e132.
[27]Nyerges Á, Csörgő B, Nagy I, et al. A highly precise and portable genome engineering method allows comparison of mutational effects across bacterial species[J]. Proc Natl Acad Sci USA, 2016, 113(9):2502-2507.
[28]Dalia AB, McDonough EK, Camilli A. Multiplex genome editing by natural transformation[J]. Proc Natl Acad Sci USA, 2014, 111(24):8937-8942.
[29]Warner JR, Reeder PJ, Karimpour-Fard A, et al. Rapid profiling of a microbial genome using mixtures of barcoded oligonucleotides[J]. Nat Biotechnol, 2010, 28(8):856-862.
[30]Sandoval NR, Kim JYH, Glebes TY, et al. Strategy for directing combinatorial genome engineering inEscherichia coli[J]. Proc Natl Acad Sci USA, 2012, 109(26):10540-10545.
[31]Jiang W, Bikard D, Cox D, et al. RNA-guided editing of bacterial genomes using CRISPR-Cas systems[J]. Nat Biotechnol, 2013,31(3):233-239.
[32]Standage-Beier K, Zhang Q, Wang X. Targeted large-scale deletion of bacterial genomes using CRISPR-nickases[J]. ACS Synthetic Biology, 2015, 4(11):1217-1225.
[33]Ronda C, Pedersen LE, Sommer MOA, et al. CRMAGE:CRISPR optimized mage recombineering[J]. Sci Rep, 2016, 6(19452):1-11.
[34]Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPRCas systems[J]. Science, 2013, 339(6121):819-823.
[35]Esvelt KM, Mali P, Braff JL, et al. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing[J]. Nature Methods,2013, 10(11):1116-1121.
[36]Krishnakumar R, Grose C, Haft DH, et al. Simultaneous noncontiguous deletions using large synthetic DNA and site-specific recombinases[J]. Nucleic Acids Research, 2014, 42(14):e111-e111.
[37]Pósfai G, Plunkett G, Fehér T, et al. Emergent properties of reduced-genomeEscherichia coli[J]. Science, 2006, 312(5776):1044-1046.
[38]Borrel G, McCann A, Deane J, et al. Genomics and metagenomics of trimethylamine-utilizing Archaea in the human gut microbiome[J]. The ISME Journal, 2017, 2017((11)):2059-2074.
[39]Ostrov N, Landon M, Guell M, et al. Design, synthesis, and testing toward a 57-codon genome[J]. Science, 2016, 353(6301):819-822.
[40]刘夺, 杜瑾, 赵广荣, 等. 合成生物学在医药及能源领域的应用[J]. 化工学报 , 2011, 62(9):2391-2397.
[41]杜若曦, 郭明璋, 谢子鑫, 等. 合成生物学在改善肠道健康状态中的应用与展望[J]. 生物技术通报, 2018, 34(1):49-59.
