基于线粒体16S rRNA全序列分析我国绵羊痒螨兔亚种的遗传多样性
2018-06-07李燕芳赵习彬赖为民杨光友古小彬
古 江,李燕芳,赵习彬,谢 跃,赖为民,杨光友,古小彬
(四川农业大学动物医学院,成都 611130)
中国作为世界最大养兔出产国之一, 26个省(自治区、直辖市)家兔总出栏量5亿2 000万只,总存栏量2亿2 400万只(2015年兔产业体系数据统计)[1];饲养的家兔种类包括肉兔、獭兔和毛兔,其中肉兔和獭兔存栏量和出栏量分别占全国的91.58%和99.80%[2];2015年我国兔肉产量87.4万t[1],兔皮约1.1亿多张,兔毛产量约2万t[3],兔皮、兔毛和兔肉总产量和出口量多年稳居世界第一。由此可见,养兔业在我国畜牧业经济中占有举足轻重的地位。
痒螨病是家兔养殖中常见的外寄生虫病之一,据报道我国家兔感染率可达70.0%[4]。该病是由绵羊痒螨兔亚种(Psoroptesovisvar.cuniculi,又名兔痒螨)寄生于家兔的外耳道皮肤表面所引起,表现出瘙痒,挠耳,外耳道内卷纸样结痂为主要临床症状,造成家兔体重减轻、饲料转化率下降、免疫功能降低,严重者出现死亡,给养兔业造成巨大的经济损失[5-7]。物种的遗传多样性可受环境和地理隔离等因素影响而出现变异,这点已在日本血吸虫[8]、捻转血矛线虫[9]、湖北钉螺[10]等物种中被证实。我国家兔饲养区域范围广泛、饲养品种较多,家兔品种和饲养区域的地理隔离是否会对家兔体表寄生的痒螨产生遗传变异?相关研究目前仍十分缺乏。线粒体DNA (mitochondrial DNA,mtDNA)具备母系遗传和高突变等特征,常作为研究物种进化的遗传标记[11-13]。我们课题组前期基于线粒体的编码蛋白基因Cytb基因对我国绵羊痒螨兔亚种的遗传变异进行了分析[14],但因基因间存在不平衡的进化关系,仅选用一个线粒体基因进行遗传变异分析并不能准确地反映种群间的进化[15]。因此,本研究中我们选取了线粒体基因中非编码蛋白基因——16S rRNA基因再次分析我国家兔主要养殖区域的肉兔和獭兔体表寄生的绵羊痒螨兔亚种的遗传多样性特征,以期得到更为准确的我国绵羊痒螨兔亚种种群遗传多样性信息,从而为养兔业中痒螨病的防控提供基础资料。
1 材料与方法
1.1 实验材料
从我国5个地理区域(华北、华东、华中、西北和西南)采集自然感染痒螨的肉兔和獭兔的外耳道结痂,从结痂中分离螨虫经形态学鉴定为绵羊痒螨兔亚种[16],于-20 ℃保存备用。其中分离自獭兔的痒螨样品25个,分离自肉兔的痒螨样品58个,共计83个样品(表1)。
1.2 DNA的提取、PCR扩增和产物测序
将“1.1”中采集的各地痒螨样品随机挑选单个痒螨进行无菌研磨后,按照OMEGA公司的节肢动物DNA提取试剂盒(DNA Mollusc Kit)的说明书提取单个螨虫的基因组DNA,提取的DNA保存于-20 ℃备用。根据GenBank上已发表的绵羊痒螨线粒体基因组序列 (登录号:KJ957822)[12]设计特异性引物,扩增16S rRNA基因全序列。上游引物F-5′-AGGTATCGGAAGGTGCCTCT-3′;下游引物:R-5′-TCCCCTACCCCTAAAAGCTCA-3′。PCR扩增体系为25 μL:4 μL模板DNA,上、下引物各1 μL,6.5 μL ddH2O,12.5 μL 2 ×TaqPCR Master Mix。PCR反应条件:94 ℃ 5 min;94 ℃ 30 s,58 ℃ 45 s,72 ℃ 1 min,35个循环;72 ℃ 10 min。阴性对照模板使用无菌双蒸水。PCR反应结束后,取5 μL PCR扩增产物进行1%的琼脂凝胶电泳检测目的片段的大小,随后将PCR阳性产物送至生物工程(上海)股份有限公司进行正反双向测序,以保证测序结果的准确性。
1.3 数据处理及分析
将测序结果按照峰图人工核对,运用BLASTn和DNAMAN 6.0.3软件对序列进行比对、拼接、剪切,构成对应的16S rRNA全序列文件。运用MEGA 5.0软件对比核苷酸序列以及对碱基百分比和种群间遗传距离进行分析;采用DnaSP 5.0软件对变异位点数、单倍型数、遗传分化系数(Fst)、核苷酸多样性(Pi)和单倍型多样性(Hd)进行统计,并计算基因流Nm=1/4(1/Fst-1);采用Arlequin 3.1软件分析种群间的遗传差异(Fst)和分子变异(AMOVA)以了解不同地区的遗传结构,并对各种群进行中性检验(Tajima’sD和Fu’sFs);构建单倍型网络分布图和采用NJ法构建系统发育树,并用bootstraps(重复1 000次)检验聚类树的置信度。
2 结 果
2.1 核苷酸序列分析
成功获得83个绵羊痒螨兔亚种样品的16S rRNA全序列,经分析获得的83条16S rRNA序列,全长均为1 025 bp(GenBank No.:MH 304762~MH 304844)。DnaSP 5.0软件分析显示83条基因序列中共有83个变异位点,其中包括68个单变异位点(81.93%)和15个简约性位点(18.07%),无碱基缺失和插入,其中转换突变的数量大于颠换突变数量。
2.2 单倍型分布
83条序列共存在52个单倍型(H1~H52),其中H2、H4、H9、H16、H32为共享单倍型;H4单倍型共享频率最高。单倍型H2由华北地区的2个样本(HB2与HB4)与西南地区的9个样本所共享(XN2、XN8、XN12、XN31、XN33~35、XN42、XN43);单倍型H4由华北地区(HB5)、华中地区(HZ1)、西北地区(XB1、XB6、XB7)、西南地区(XN10、XN43、XN44、XN45、XN46、XN47、XN48)共享;单倍型H9由华东地区(HD2、HD3、HD8)、西南地区(XN20、XN23、XN25、XN26)共享;单倍型H16由华中地区(HZ4、HZ6、HZ8、HZ9)、西南地区(XN19)共享;单倍型H32由XN21、XN22共享;其余的均是各种群地区特有的单倍型(表1)。
2.3 单倍型多样性及核苷酸多样性
5个地理种群的单倍型多样性(Hd)为0.857 14(西北地区)~0.964 29(华北地区),我国整体种群的单倍型多样性(Hd)较高(0.958 27),表明我国整体种群及各地种群的绵羊痒螨种群单倍型多样性(Hd)较为丰富(表1)。我国整体种群的平均核苷酸多样性(Pi)为0.006 86,而5个地理种群中,以华中地区的核苷酸多样性(Pi)最低(0.001 71),华东地区的核苷酸多样性(Pi)最高(0.006 76),表明我国这5个区域的核苷酸多样性(Pi)较高;5个地理种群核苷酸差异指数(K)的范围在1.755 56(华中地区)~6.928 57(华东地区)(表1)。
2.4 遗传距离、中性检验值和遗传分化指数
5个地理种群中,除华北地区的Tajima’sD检验值为正值(0.067 96),其余4个地理种群均为负值(-1.658 34~-0.959 56),西北地区Tajima’sD绝对值最高,华北地区Tajima’sD绝对值最低,并且华中地区、西北地区、西南地区种群差异显著(P<0.005)。除华东地区的Fu’sFs值为正外(0.183 69),其余4个区域值均为负(-20.424 13~-0.003 85),以西南地区绝对值最高,西北地区绝对值最低,华中地区和西南地区种群差异显著(P<0.005)(表1)。
华北地区与华东地区、华中地区、西北地区、西南地区的遗传距离分别为0.008、0.005、0.005、0.007,且差异显著(P<0.005);华东地区与华中地区、西北地区、西南地区的遗传距离分别为0.010、0.009、0.006,且差异显著(P<0.005);华中地区与西北地区、西南地区的遗传距离分别为0.004和0.009,且差异显著(P<0.005);西北地区与西南地区的遗传距离为0.008,遗传距离差异显著(P<0.005)(表2)。
5个地理种群间的Fst值(-0.037 59~0.587 63)和基因流值-6.900 7~31.117 62变化较大(表3),其中华中地区与华东地区的Fst值最高,并且遗传分化差异显著(P<0.005),这两个区域的基因流Nm为0.175 43,表明种群间存在较小的基因流;华东地区与西南地区基因流值Nm(31.117 62)最高,表明种群间基因交流频繁,导致两者间的遗传分化差异不显著(Fst值=0.007 97,P>0.005)。
2.5 系统发育树与单倍型网络图的构建
NJ树显示(图1),绵羊痒螨兔亚种的52个单倍型构成2个大的分支。2大分支包含了不同的地理来源及温度带的单倍型,单倍型的分布无规律可循,尚未形成显著的地理种群结构和温度种群结构。
单倍型网络图显示(图2),单倍型被分成两部分,各单倍型呈发散分布,相邻单倍型之间的突变从1步到5步不等,网络图中各单倍型的聚类情况与NJ树的聚类情况非常相似,同样未形成显著的地理种群结构。
3 讨 论
本研究首次成功地利用PCR技术扩增了我国5大区域的83株绵羊痒螨兔亚种的16S rRNA全序列,并分析了其遗传多样性和种群结构特征。我们采用的单个痒螨虫体进行DNA提取,克服了多个痒螨虫体提取DNA所带来的不准确性,避免了遗传变异的不准确性[17-19]。研究表明地理环境和宿主的改变会影响寄生虫种群遗传结构[20],前期我们课题组基于Cytb分析亦证实我国地理环境差异较大的各地采集的绵羊痒螨间存在较高的遗传多样性,有一定的遗传分化,但未分化形成地理种群结构[14]。但基于单个基因得到的遗传变异结果并不一定能准确地反映种群间的进化关系,因此,本研究采用线粒体16S rRNA基因进一步分析我国各地绵羊痒螨的遗传多样性。
环境的改变会导致动物物种自身变异的产生,遗传变异的大小表现出其遗传多样性的高低,遗传多样性低的物种种群很难在多变的环境中生存。所以遗传多样性较高的物种比变异较低的物种更具备进化潜力和环境适应性,这更有利于种群的发展[21]。通常用单倍型多样性(Hd)和核苷酸多样性(Pi)来衡量一个物种或群体遗传多样性。本研究中83个样本共有52个单倍型,其Hd达0.958 27,表明我国这5个区域的绵羊痒螨的单倍型多样性较为丰富。然而本研究中5个区域总的核苷酸多样性为0.006 86,表现出较低的核苷酸多样性水平。这种高Hd值、低Pi值的现象反映了绵羊痒螨种群在历史中可能出现过瓶颈效应而出现种群扩张,种群快速的自身变异以致其较高单倍型多样性,但并没有积累较高的核苷酸多样性水平[22]。这种高Hd、低Pi的现象常发现于大多数具有较大的母系有效种群和较强繁殖能力的无脊椎动物中[23-24]。再者,中性检验(Tajima’D和Fu’sFs)结果亦证明了我国绵羊痒螨群体在历史上可能出现过种群扩张现象,从而易于该种群的生存。
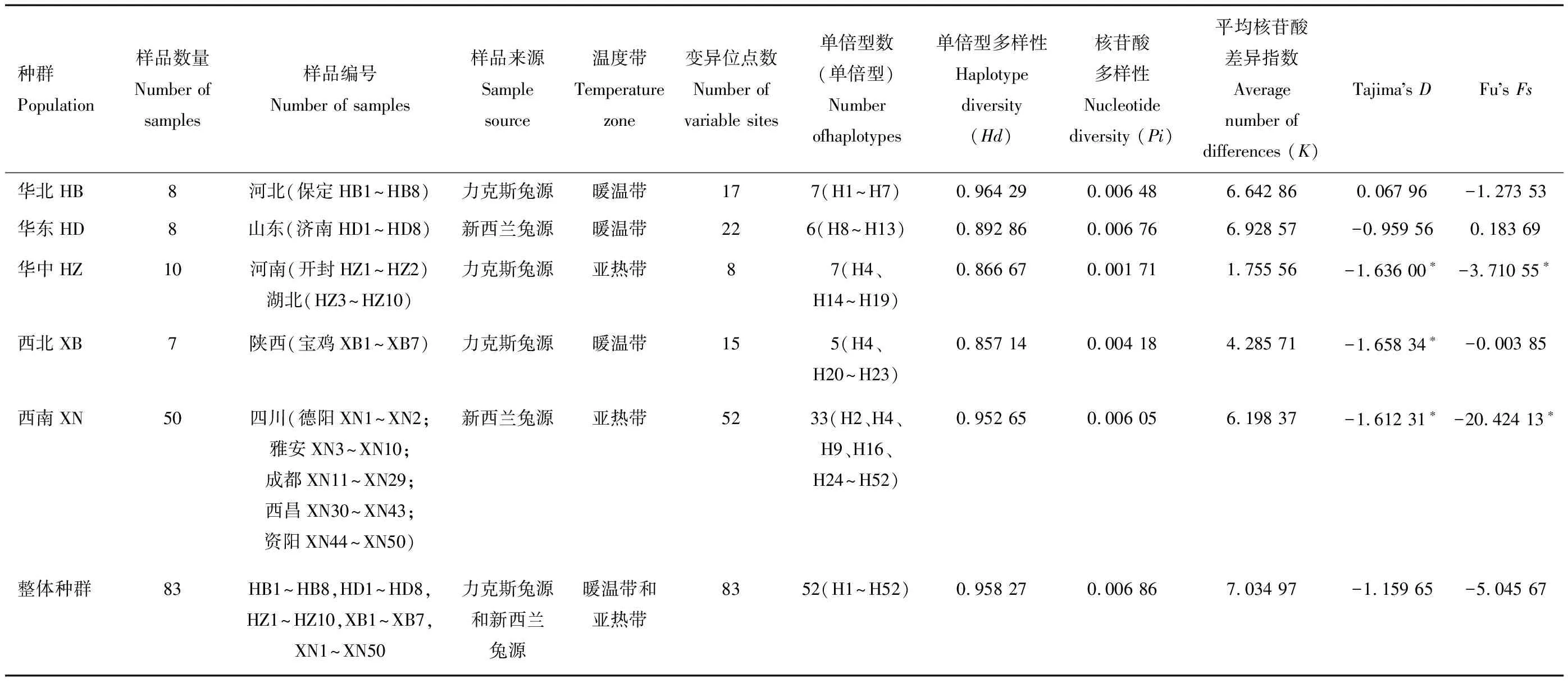
表1 基于16S rRNA分析我国5个地理区域绵羊痒螨兔亚种种群的遗传多样性指数Table 1 Indexes of genetic diversity of Psoroptes ovis var. cuniculi populations from five regions in China, based on 16S rRNA
*.P<0.005,表示差异显著
*.P<0.005, indicates significant difference

表2 我国5个地理区域绵羊痒螨兔亚种种群的遗传距离Table 2 Genetic distance of Psoroptes ovis var. cuniculi populations between five region populations in China

表3 我国绵羊痒螨兔亚种种群的基因流(Nm)(上三角)和遗传分化度(Fst)(下三角)Table 3 Gene flow (Nm) (above diagonal) and genetic diversity (Fst) (below diagonal) between Psoroptes ovis var. cuniculi populations in China
*.P<0.005,表示差异显著
*.P<0.005, indicates significant difference
本次研究的5个地理区域跨越了亚热带和暖温带,同时被我国的南北分界线——秦岭-淮河所隔离以及长江、黄河等河流的阻断,这些天然的地理屏障形成了物种的地理隔离,地理的隔离可能会造成生殖隔离,因此天然的地理屏障形成了我国绵羊痒螨兔亚种种群之间的基因交流障碍。但从反映物种遗传分化强弱的基因流(Nm)和遗传分化系数(Fst)来看[25],华东(山东)与西南(四川)种群间基因流值>4(Nm=31.117 62),Fst值<0.05(Fst=0.007 97),表明2个群体间基因交流充分,种群出现遗传分化可能性小[25];而华北与华东、西南以及华中与西北种群间亦存在一定的基因交流(Nm>1),种群间遗传分化较大(0.15

HB.华北;HD.华东;HZ.华中;XB.西北;XN.西南HB. Huabei;HD. Huadong;HZ. Huazhong;XB. Xibei;XN. Xi’nan图1 基于痒螨16S rRNA的52个单倍型构建的NJ树Fig.1 NJ Tree based on52 haplotypes of 16S rRNA of Psoroptes ovis var. cuniculi
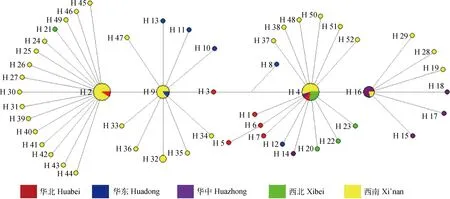
单倍型的分布频率与圆圈的大小成正比;不同颜色表示不同样品来源The haplotype distribution frequency is proportional to the size of the circle; Different colors represent different sample sources图2 基于16S rRNA构建的绵羊痒螨单倍型网络图Fig.2 A network map of Psoroptes ovis, based on 16S rRNA
系统发育树(NJ树)和单倍型网络图显示我国绵羊痒螨16S rRNA单倍型构成2大分支,呈现出杂乱的分布格局,绵羊痒螨兔亚种虫株并未按照地理来源聚集,同时亦未根据宿主(獭兔还是肉兔)而进行聚集,还未根据是源自亚热带还是暖温带而聚集(图1和图2),未形成显著的地理种群结构、温度带种群结构和兔品种种群结构。从遗传距离、基因流及遗传分化指数来看,处于亚热带的西南地区与处于温带的华东西区间基因流值较大,遗传分化指数较小(表2和表3)。因此,我国绵羊痒螨种群间的基因交流与地理、兔品种和温度带无关。
4 结 论
本研究首次成功地利用PCR技术扩增了我国5大区域的83株单个绵羊痒螨兔亚种的线粒体16S rRNA全序列,分析了5个地理区域绵羊痒螨兔亚种的遗传多样性和种群结构特征。发现我国绵羊痒螨兔亚种的遗传多样性较高,遗传分化不明显,尚未形成地理种群结构。
参考文献(References):
[1] 何经纬, 徐 旭, 关云秀, 等. 四川兔业发展现状及建议[J]. 四川畜牧兽医, 2016, 43(11): 7-9.
HE J W, XU X, GUAN Y X, et al. Current status and suggestions for future development of rabbit industry in Sichuan[J].SichuanAnimal&VeterinarySciences, 2016, 43(11): 7-9. (in Chinese)
[2] 刘汉中, 秦应和, 武拉平, 等. 中国兔产业发展现状及“十三五”科技创新重点思考[J]. 中国养兔, 2016(6): 32-34.
LIU H Z, QIN Y H, WU L P, et al. Rabbit industry in China: Current status and thinking about technology innovation in the 13th national five-year plan[J].ChineseJournalofRabbitFarming,2016(6): 32-34. (in Chinese)
[3] 孙海涛, 张 印, 白丽雅, 等. 山东养兔发展现状及对策[J]. 山东畜牧兽医, 2016, 37(9): 75-76.
SUN H T, ZHANG Y, BAI L Y,et al. Current status and innovative solutions of rabbit in Shandong province[J].ShandongJournalofAnimalScienceandVeterinaryMedicine, 2016, 37(9): 75-76. (in Chinese)
[4] 柏克仁. 民和县甘沟乡兔痒螨病调查[J]. 青海畜牧兽医杂志,2012, 42(3): 12.
BAI K R.Survey on psoroptic mange of rabbit in Min-he county of Gangou country[J].ChineseQinghaiJournalofAnimalandVeterinarySciences, 2012, 42(3): 12. (in Chinese)
[5] ULUTAS B, VOYVODA H, BAYRAMLI G, et al. Efficacy of topical administration of eprinomectin for treatment of ear mite infestation in six rabbits[J].VetDermatol,2005, 16(5): 334-337.
[7] SHANG X F, WANG D S, MIAO X L, et al. The oxidative status and inflammatory level of the peripheral blood of rabbits infested withPsoroptescuniculi[J].ParasitVectors,2013, 7: 124.
[8] 郭凯文, 牛安欧. 日本血吸虫线粒体DNA两个分子的遗传变异[J]. 中国寄生虫学与寄生虫病杂志, 2004, 22(5): 300-302.
GUO K W, NIU A O. Studies on the genetic variation of two mitochondrial DNA molecules ofSchistosomajaponicum[J].ChineseJournalofParasitologyandParasiticDiseases, 2004, 22(5): 300-302. (in Chinese)
[9] YIN F Y, GASSER R B, LI F C, et al. Genetic variability within and amongHaemonchuscontortusisolates from goats and sheep in China[J].ParasitVectors,2013, 6: 279.
[10] DAVIS G M, WU W P, XU X J. Ecogenetics of shell sculpture inOncomelania(Gastropoda) in canals of Hubei, China, and relevance for schistosome transmission[J].Malacologia, 2006, 48(1-2): 253-264.
[11] OKAMOTO M, BESSHO Y, KAMIYA M, et al. Phylogenetic relationships withinTaeniataeniaeformisvariants and other taeniid cestodes inferred from the nucleotide sequence of the cytochrome c oxidase subunit I gene[J].ParasitolRes, 1995, 81(6): 451-458.
[12] BART J M, BARDONNET K, ELFEGOUN M C B, et al.Echinococcusgranulosusstrain typing in North Africa: Comparison of eight nuclear and mitochondrial DNA fragments[J].Parasitology, 2004, 128(2): 229-234.
[13] ZHANG L H, JOSHI D D, MCMANUS D P. Three genotypes ofEchinococcusgranulosusidentified in Nepal using mitochondrial DNA markers[J].TransRSocTropMedHyg, 2000, 94(3): 258-260.
[14] 赵习彬, 王保健, 简克灵, 等. 基于线粒体细胞色素氧化酶b(cytb)全基因序列分析我国部分地区兔痒螨的遗传变异特征[J]. 畜牧兽医学报, 2017, 48(4): 714-721.
ZHAO X B, WANG B J, JIAN K L, et al.Analysis of the characteristics of genetic variability withinPsoroptescuniculiisolates in some regions of China, inferred by mitochondrial cytochrome oxidase b gene[J].ActaVterinariaetZootechnicaSinica,2017, 48(4): 714-721. (in Chinese)
[15] ZHAO Y E, CAO Z G, CHENG J, et al. Population identification ofSarcopteshominisandSarcoptescanisin China using DNA sequences[J].ParasitolRes, 2015, 114(3): 1001-1010.
[16] SWEATMAN G K. On the life history and validity of the species inPsoroptes, a genus of mange mites[J].CanJZool, 1958, 36(6): 905-929.
[17] GU X B, LIU G H, SONG H Q, et al. The complete mitochondrial genome of the scab mitePsoroptescuniculi(Arthropoda: Arachnida) provides insights into Acari phylogeny[J].ParasitVectors, 2014, 7: 340.
[18] ALASAAD S, ROSSI L, MAIONE S, et al. HotShot Plus ThermalSHOCK, a new and efficient technique for preparation of PCR-quality mite genomic DNA[J].ParasitolRes, 2008, 103(6): 1455-1457.
[19] SASTRE N, RAVERA I, FERREIRA D, et al. Development of a PCR technique specific forDemodexinjaiin biological specimens[J].ParasitolRes, 2013, 112(9): 3369-3372.
[20] BARRETT L G, THRALL P H, BURDON J J, et al. Life history determines genetic structure and evolutionary potential of host-parasite interactions[J].TrendsEcolEvol, 2008, 23(12): 678-685.
[21] 王 凝, 古小彬, 汪 涛, 等. 基于cox1基因对中国青藏高原地区细粒棘球绦虫遗传多态性的研究[J]. 畜牧兽医学报, 2015, 46(3): 453-460.
WANG N, GU X B, WANG T, et al.Genetic variability ofEchinococcusgranulosusdetermined by the mitochondrial cytochrome c oxidase subunit 1 gene in the Tibet plateau of China[J].ActaVterinariaetZootechnicaSinica,2015, 46(3): 453-460. (in Chinese)
[22] AVISE J C. Phylogeography: the history and formation of species[M]. Cambridge: Harvard University Press, 2000.
[23] GRANT W A S, BOWEN B W. Shallow population histories in deep evolutionary lineages of marine fishes: Insights from sardines and anchovies and lessons for conservation[J].JHered, 1998, 89(5): 415-426.
[24] LAVERY S C, MORITZ C, FIELDER D R. Indo-Pacific population structure and evolutionary history of the coconut crabBirguslatro[J].MolEcol,1996, 5(4): 557-570.
[25] 郝桂英, 杨应东, 杨光友. 四川省山羊多头蚴线粒体cox2基因序列测定及种系发育分析[J]. 中国动物传染病学报, 2015, 23(1): 54-59.
HAO G Y, YANG Y D, YANG G Y. Sequencing and phylogenetic analysis of mitochondrialcox2 gene ofCoenurusin goats in Sichuan province[J].ChineseJournalofAnimalInfectiousDiseases,2015, 23(1): 54-59. (in Chinese)
[26] BLOUIN M S, YOWELL C A, COURTNEY C H, et al. Host movement and the genetic structure of populations of parasitic nematodes[J].Gentics, 1995, 141(3): 1007-1014.
[27] ARAYA-ANCHETTA A, BUSCH J D, SCOLES G A, et al. Thirty years of tick population genetics: A comprehensive review[J].InfectGenetEvol, 2015, 29: 164-179.
