上海白猪(上系)基因组遗传变异检测与功能注释分析
2018-06-07盛中华陈国宏潘玉春王起山
盛中华,陈国宏,潘玉春,王起山,张 哲
(1.扬州大学动物科学与技术学院,扬州 225009;2.上海市闵行区动物疫病预防控制中心,上海 201109;3.上海交通大学农业与生物学院,上海 200240)
上海白猪是通过十几年的选育而培育成的一个地方肉脂兼用型品种,具有肉质好、产仔数较多、胴体瘦肉率较高和耐粗饲等优良特点。早在1986年被收录至《中国猪品种志》, 2012年列入《上海市畜禽遗传资源保护名录》。上海白猪(上系)作为上海白猪的一个品系,曾作为供港猪杜长上杂交组合的母本风靡一时,但随着引进的国外猪种严重地冲击国内种猪市场,上海白猪(上系)的群体规模受到了极大的影响,各项生产指标出现一定程度的衰退,因此需要深入了解当前群体的遗传现状,以便更好地进行提纯复壮与开发利用。
遗传变异的检测和功能注释是分析动物群体遗传现状的重要前提和基础,单核苷酸多态性(SNP)和2 bp~1 kb 之间的小片段插入和缺失(InDel)为猪基因组占比最大的两类分子标记。其中,SNP因具备充足的信息常用于基因连锁分析和与动植物DNA功能性变异密切相关的连锁不平衡分析。如H.S.Ai等[1-2]基于全基因组的SNP遗传标记开展了中国部分地方猪的群体结构和连锁不平衡分析等。M.Z.Li等[3]利用重测序得到的SNP标记,从群体遗传学上进一步阐明了藏猪适应高原环境的遗传机理及杜洛克经人工选择后与藏猪遗传上的差异。InDel标记具有较好的稳定性和多态性等优点,频率仅次于SNP,其中约1/3位于已知的基因区域内,还有一些位于决定基因功能的关键性区域如启动子区和外显子区,当前已有多个研究组利用基因芯片开展猪抗病、肉质等相关遗传机制研究[4-6]。Q.Xiao等[7-9]也基于基因组简化测序技术(GGRS)开展了地方猪和引进猪的SNP和InDel的检测分析。
本研究针对上海白猪核心群体,采用GGRS测序技术[10-11],在全基因组范围内进行SNP和InDel等遗传变异多态性检测,并进行系统的功能注释分析,以便了解上海白猪当前群体的遗传现状,进而为其保护和利用提供分子生物学依据。
1 材料与方法
1.1 上海白猪及对照群体
试验所用99头上海白猪的耳组织样采自上海市闵行区畜禽种场的保种群。同时,为了更准确的开展遗传变异的检测分析和缺失基因型的填补,揭示上海白猪的种质特性,本研究采用与其地理位置邻近的中国地方品种(包括6个太湖流域地方品种:梅山猪、二花脸猪、枫泾猪、米猪、沙乌头、嘉兴黑猪)及西方引进品种(包括杜洛克、长白、大白、皮特兰、巴克夏)作为对照品种,共计447个个体。
1.2 简化基因组测序
上海白猪采样个体基于Illumina公司Hiseq2000平台,采用基因组简化测序技术进行双端测序[10-11]。为了更好的与其它品种进行比较,将上海白猪的测序结果与太湖猪和引进猪种11个品种测序结果[12-14]在一起进行SNP 和InDel calling,缺失基因型的填补则利用Beagle 4.1来完成[15-16]。最后,分别在全群和上海白猪群过滤掉最小等位基因频率(MAF)小于0.05的SNP位点,用于后续分析。
1.3 基因功能区间分布
猪的基因注释数据来自Ensembl数据库(Ensembl release 78,ftp://ftp.ensembl.org/pub/release-78/gtf/sus_scrofa/)[17-18],共包含25 332个基因注释信息。根据数据库的信息,本研究主要将基因组区间分为起始或者终止密码子、外显子、内含子、非翻译区(UTR)和基因间区。利用PERL脚本编程语言分析SNP、InDel是否在上述基因组功能区间。
1.4 基因功能富集分析
位于起始/终止密码子区或者外显子区的变异很有可能改变基因的结构和功能。因此,把位于起始/终止密码子区或者外显子区的变异定义为大效应突变。为了进一步了解各类大效应突变显著参与的通路、生物学功能和参与的分子生物学过程,利用DAVID v6.7网络数据库工具及GO和KEGG数据库[19-20],对这些与大效应突变相关的基因,按突变类型分别进行功能注释(GO annotation)和通路分析(Pathway analysis)。显著性水平P阈值设定为0.05[21-22]。针对当前猪的GO与KEGG注释库还很不全面的问题,利用PERL脚本提取猪对应的人同源基因标识用于基因富集分析。
1.5 与QTL的映射
猪许多重要性状的QTL已相继被定位,并被收集到猪的QTL数据库(http://www.animalgenome.org/, Release 32, Apr 27, 2017)[23-24]。截至本研究进行前,该数据库共收集了12 618 QTLs,基于现有的QTL数据库对SNP和InDel等遗传变异进行功能注释分析。针对有些QTLs长度过长,并不能有效地用于后续分析问题,本研究去除了长度超过1 Mb的QTLs,并且把重叠50%以上的两个QTLs合并成一个新QTL,同时设定新QTL与其原始性状都相关。此过程利用QTL的位置信息与性状信息,通过Perl语言脚本处理完成。
2 结 果
2.1 上海白猪(上系)基因组碱基测序质量、覆盖度和深度
经过污染序列的过滤之后,上海白猪共获得大约430万条高质量的reads,两端碱基平均质量得分均在Q30以上,即碱基测得的正确率大于99.9%。其中,最小检测个体和最大检测个体测序reads数目分别为200万条和616万条,个体平均reads数目为409万条(表1)。
测序覆盖度是指每个个体测到的基因组总碱基数占该物种基因组总碱基数的百分比,由表1可知,每个群体个体的平均覆盖度范围从1.2%(枫泾)到3.98%(长白),所有个体的平均测序覆盖度为2.46%,上海白猪的平均测序覆盖度为2.87%。测序深度是指基因组上的同一位置被测到的次数。测序深度的计算方法为每个个体测到的高质量reads数与唯一位置reads数的比值,在本研究中,上海白猪的平均测序深度为3.90,所有个体的平均测序深度为4.78倍,详见表1。
2.2 上海白猪(上系)基因组变异检测及在染色体上的分布
经SNP calling和过滤后,全群共检测到487 323个SNPs、976 235个InDels。其中,上海白猪共检测到328 586个SNPs、693 220个InDels(表2)。我们进一步统计了上海白猪各类变异在染色体上的数量和密度分布,发现序列的变异数量一般与染色体的长度相关,染色体长度越长其所含有的变异的数量越多。如表2所示,1号染色上的数量最多,SNPs和InDels分别为29 857和68 997个,而Y染色体上的数量最少,SNPs和InDels分别为39和100个。SNP和InDel在染色体上的平均密度分别为12.96/100 kb和26.67/100 kb。笔者进一步以400 kb为不重叠窗口,图形化展示SNP的数量在染色体上分布,可见SNP和InDel的密度在不同染色体上的分布有较大的差异,同一染色体上,分布相对均匀,具体分布可见图1和图2。
2.3 上海白猪(上系)基因组SNP和InDel的功能区间分布
当前研究结果显示,上海白猪SNP和InDel在基因内的数量分别为11 496和13 216个。基因上各类变异在每个染色体上的数量分布、对应的基因数量见表2。其次,按照基因的结构区域分类,我们进一步对各类变异做了统计分析。结果显示,SNP和InDel在各类功能基因区间的分布特点基本一致。它们在不同的基因区间的数量分布详见表3。由表3可知,绝大多数的SNP变异分布在基因间区,约占74.61%;内含子中比例次之,约为22.76%;外显子区域中仅占1.67%。和SNP的分布规律一致,InDel在基因间区的比例也最高,约为83.38%,内含子中比例次之,约为14.98%。
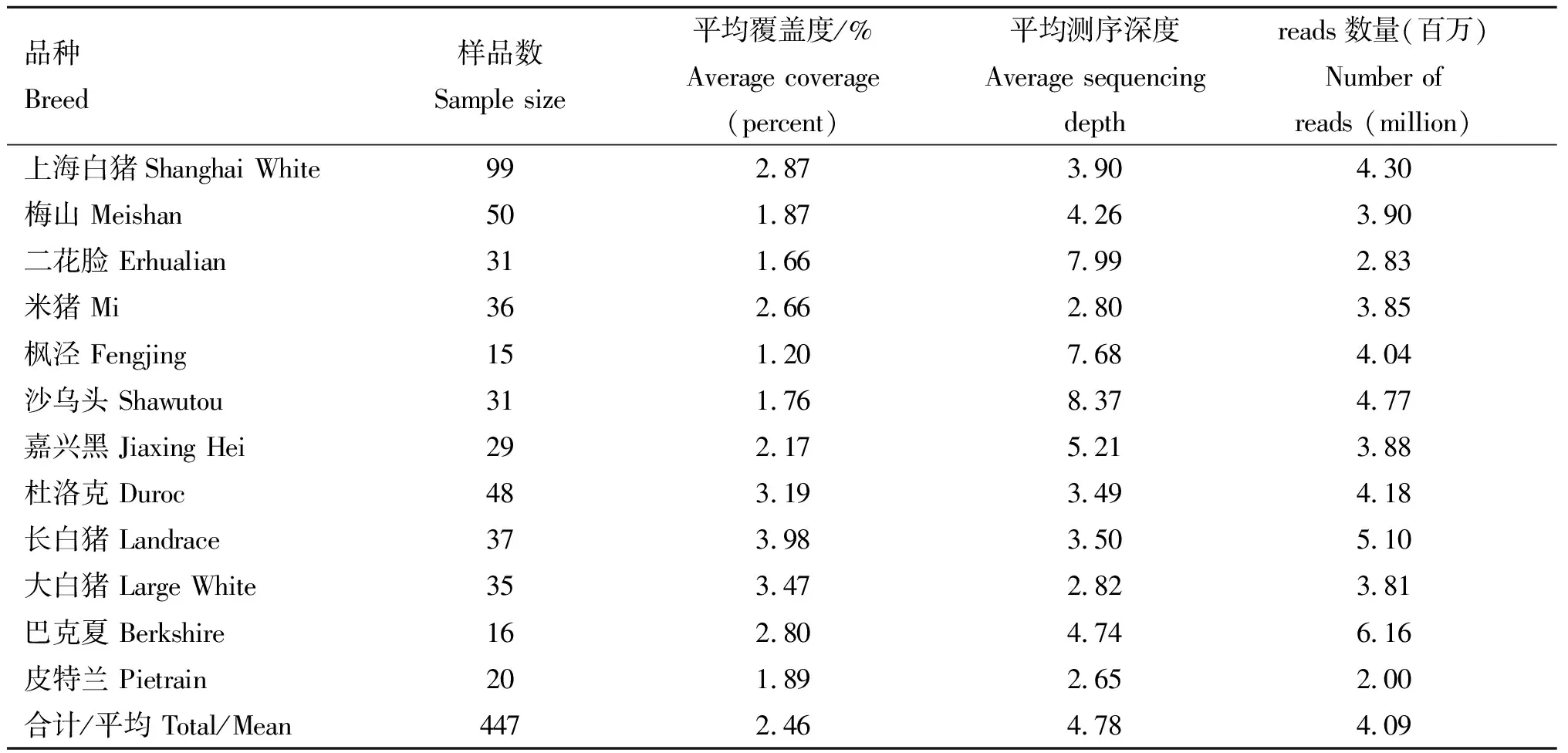
表1 测序数据量、覆盖度和深度Table 1 Distribution of high quality reads number, coverage and depth across population
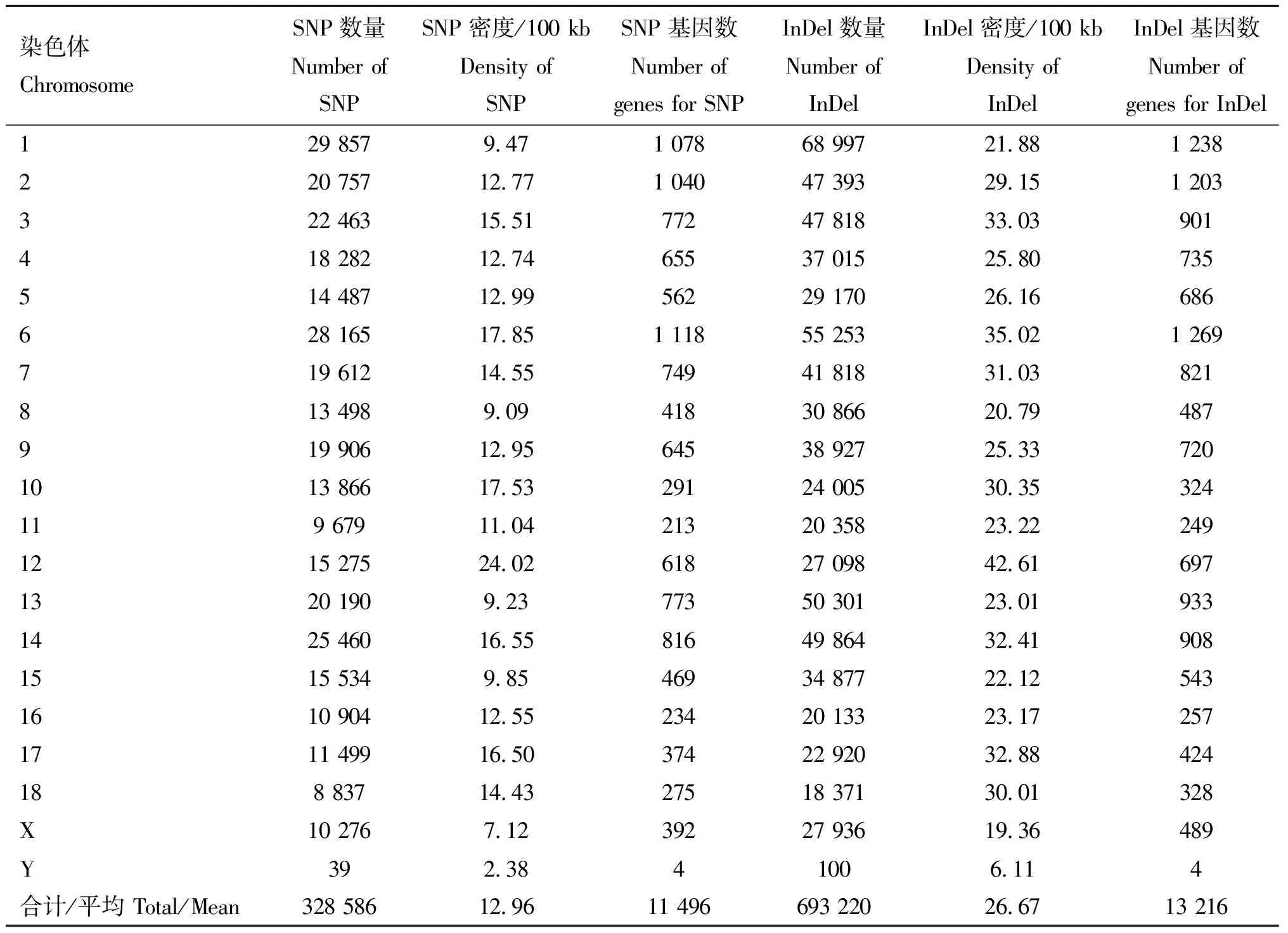
表2 SNPs和InDels遗传变异在染色体上的数量及其密度分布Table 2 The number and density distribution of SNPs and InDels detected on each chromosome

图1 SNPs在染色体上的分布Fig.1 The location distribution of SNPs across genomes

图2 InDels在染色体上的物理位置分布Fig.2 The location distribution of InDels across genome
2.4 上海白猪(上系)基因组SNP和InDel的功能注释及通路分析
通过功能注释及通路分析,分别鉴定出了上海白猪各类大效应突变对应的基因显著参与的通路和生物学过程,结果见表4。SNP、InDel显著参与的通路/生物过程数目分别为13和40个。大效应突变的基因富集分析结果显示,不同类型变异富集的通路存在差异。SNP显著参与的通路大部分是与蛋白高分子合成与调控有关。比如,蛋白代谢过程(GO:0019538)、高分子代谢过程(GO:0043170)等。而InDel多在组织、器官发育和疾病相关的通路中发生了富集,比如单细胞生物过程(GO:0044707)和致心律失常性右心室心肌病(ssc05412)等。综上,本研究结果初步表明,不同类型的变异可能影响不同的基因网络调控及生物功能。
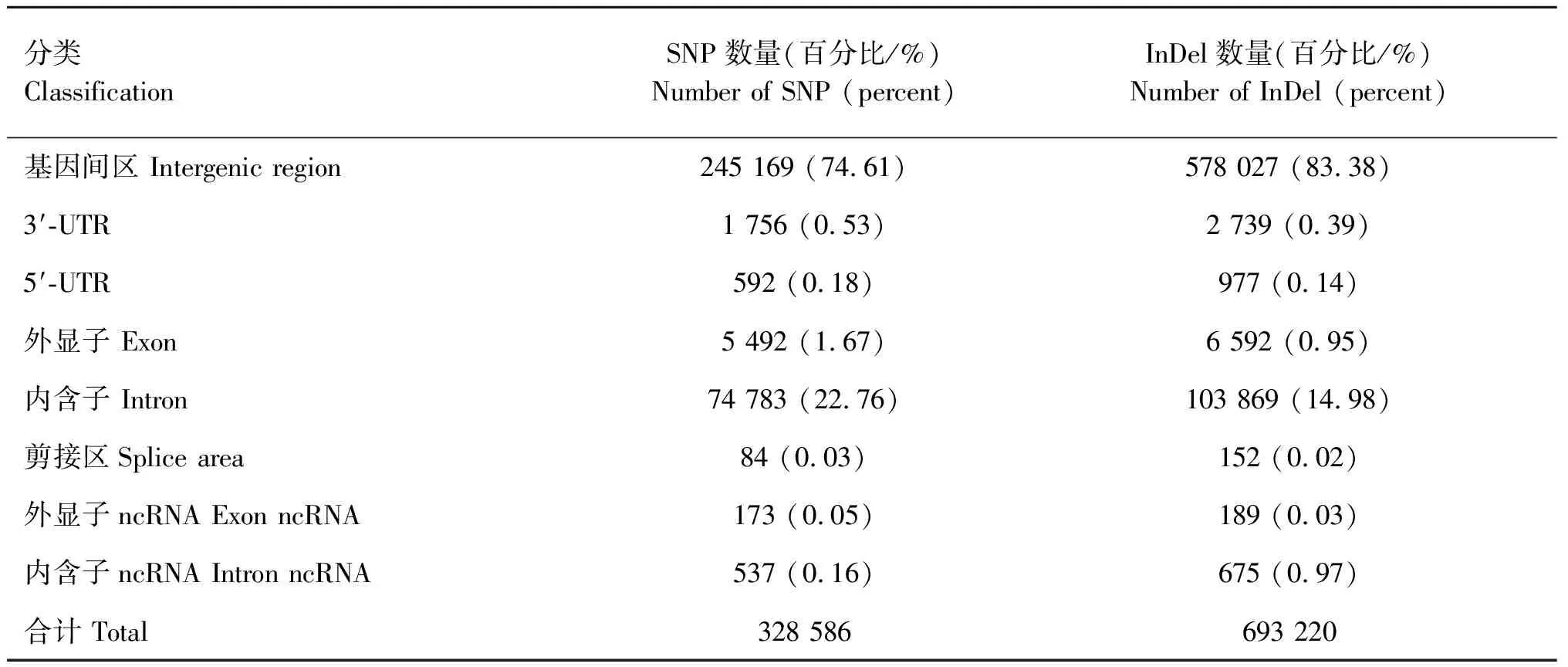
表3 SNPs和InDels在不同的基因区间的数量分布Table 3 The number distribution of SNPs and InDels across genome region

表4 各类大效应变异显著富集的GO和PathwayTable 4 Significant GO and Pathway enriched by the large-effect variants

(续表4 Continued)
(转下页Carried forward)
2.5 上海白猪(上系)基因组SNPs和InDels位于QTL的映射
经过过滤和合并之后,上海白猪SNPs和InDels在五大类QTLs注释性状中的数量分布详见表5。坐落在不同性状中的各类变异数量统计分析结果显示,SNP变异主要分布在肉质与胴体性状(256 573)和健康性状(246 230),外貌性状中最少(205 204);InDel变异在各个性状中的分布规律与SNP基本一致,肉质与酮体性状占比最多(283 936),外貌性状中最少(233 299)。
按照变异是否位于基因上,笔者又进一步开展了相关统计分析。结果显示,SNP和InDel位于基因上的数量与变异对应的基因落在QTL区间上的比例基本一致,即主要分布在肉质与酮体性状和健康性状相关的基因上,而外貌性状中最少。这表明,与肉质和健康相关的基因可能具有较高多态性,而与外貌性状相关的基因可能相对比较保守,存在的变异较低。
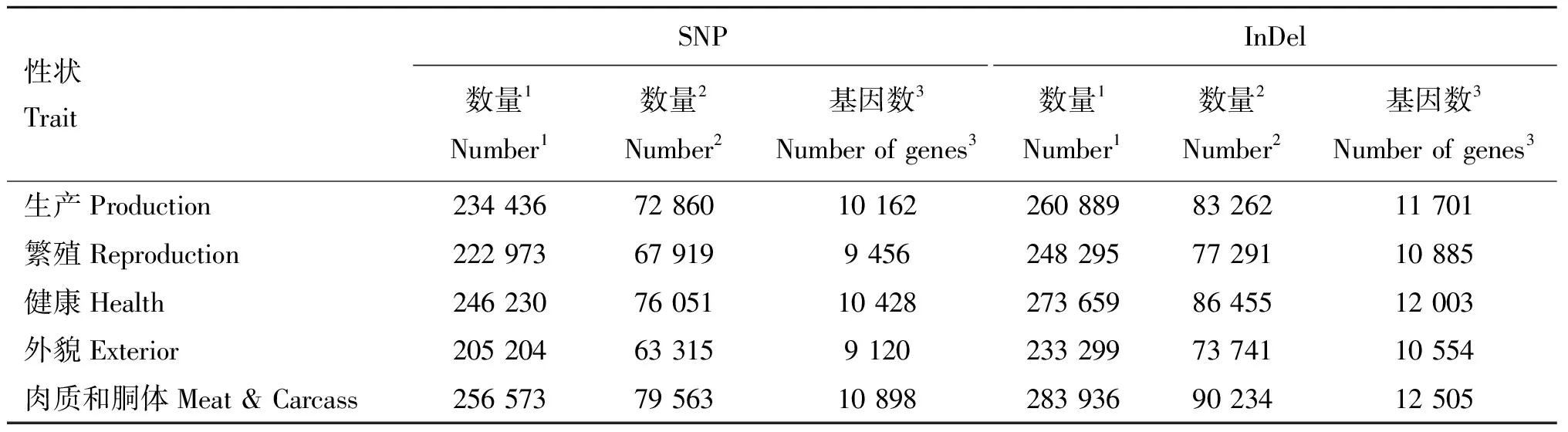
表5 同时位于QTL与基因上的各类变异数量分布Table 5 The number distribution of variants located in both QTLs and gene regions
1. 位于QTLs中的变异数量;2.位于基因上的变异数量;3.与变异对应的基因数量
1.The number of variants located in QTLs;2. The number of variants located in the genes;3. The number of genes corresponding to the variants
3 讨 论
SNP是一种重要的遗传分子标记,也是当下分子遗传学研究应用最为广泛的分子标记。在本研究中,检测到的328 586个SNPs中,有接近6%为新发现的SNPs,这表明在上海白猪中有着自己特异性,其基因组上仍然有大量的SNPs有待于被发现。通过测序技术发现,特色培育品种的遗传变异对研究中国地方猪种和培育品种都非常必要。同时,对于丰富SNP数据库也有着重要的意义。
除了进行SNP的检测外,本研究还对上海白猪InDel变异进行了检测,并发现这些变异广泛存在于上海白猪的基因组中。这些结果将有利于人们对猪基因组信息更加全面和深入的认识。通过与已有研究比较[4,7, 12, 25],本研究所检测的InDels中,26.3%也被其它研究检测到(若已报道的InDel与笔者检测的重叠区域超过任一方长度的80%,则认为是同一个)。这些研究也从另一个侧面证实了本研究检测的可靠性。
在揭示上海白猪基因组遗传分子的特征上,本研究主要对遗传变异分子在染色体上的数量、密度、物理位置等分子特征进行了分析。研究结果表明,上海白猪的遗传分子具有下列特征:第一,SNP在不同染色体上的密度分布存在差异。值得关注的是12号染色体,其变异的密度明显高于其它染色体。笔者查看了Ensembl 的基因数据库和QTLs数据库,同样发现12号染色体具有最高密度的基因分布,而且其QTL密度也在前3位。由此可以看出,12号染色体可能在基因网络和性状表现上发挥着非常重要的作用,值得研究者对此染色体重点关注。第二,除少量位置出现过密或过疏,变异在同一条染色体上的物理分布相对均一。因此,本研究检测的SNP可以作为全基因组范围内的遗传标记,用于遗传多样性、群体结构、信号选择、全因组关联分析等研究中。
此外,研究发现,很多基因受到InDel改变阅读框的影响。其中,一些基因与繁殖性状相关,例如CXCL10基因,S.Dall′olio等[26]报道,CXCL10基因可能参与胚胎的发育和着床,该基因中存在的多态变异可以作为意大利大白猪产仔数关联研究的标记。另外,一些基因是与免疫相关,如IRF7基因,IRF7基因属于干扰素调控因子家族成员,在调节Ⅰ型干扰素抵抗病原体的感染中发挥着非常关键的作用[27-28]。而IFIT1基因同时作为传递和受体分子来抵抗几类病毒家族病原体[29]。值得注意的是,基因富集分析结果也表明,InDel相关基因在多个与疾病相关的通路中发生了富集(如“ssc05414:Dilated cardiomyopathy” 和 “ssc05410:Hypertrophic cardiomyopathy (HCM)”)。因此推测,上海白猪的InDel极有可能在繁殖性状与免疫性状中发挥着重要作用。
QTL数据库包含了猪经济性状的重要遗传信息,因此对于动物遗传研究学者是一个巨大的宝藏。为了更清楚地了解各类变异可能影响的性状,建立遗传变异与性状之间可能存在的联系,本研究开展了基于QTL数据库的映射分析。同时又对各类变异在不同性状中的分布特点进行了统计分析。结果显示,各类变异在QTL上的比例分布与基因上的比例分布相近(除个别区间),这表明各类变异在不同性状QTL中基因上的分布相对均一。另外,本研究还统计了各类变异在新定义QTL中的密度,整体而言具有较高变异密度的QTL主要是与肉质与胴体及健康性状相关,这些遗传标记将能进一步用于解析为何上海白猪在肉质和抗病力上有较好表现。这些研究结果加深了人们对上海白猪基因组遗传变异分布特点的了解,为进一步深入的研究其功能和加快分子育种进程奠定了基础,也为其后续的保护和利用提供了分子生物学依据。
4 结 论
本研究利用简化基因组测序技术对肉质好、胴体瘦肉率较高和耐粗饲的上海白猪(上系)全基因组范围内的遗传变异进行了检测,并通过对太湖流域地方猪种和西方引进品种的合并比较分析,阐释了这些遗传变异的染色体分布、基因区间和功能注释特征。本研究深入分析了上海白猪(上系)群体的遗传现状,为更好地进行提纯复壮、分子设计育种与开发利用奠定基础。
参考文献(References):
[1] AI H S,HUANG L S,REN J.Genetic diversity,linkage disequilibrium and selection signatures in Chinese and western pigs revealed by genome-wide SNP markers[J].PLoSOne,2013,8(2):e56001.
[2] AI H S,FANG X D,YANG B,et al.Adaptation and possible ancient interspecies introgression in pigs identified by whole-genome sequencing[J].NatGenet,2015,47(3):217-225.
[3] LI M Z,TIAN S L,JIN L,et al.Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars[J].NatGenet,2013,45(12):1431-1438.
[4] HAN S H,SHIN K Y,LEE S S,et al.SINE indel polymorphism of AGL gene and association with growth and carcass traits in Landrace × Jeju Black pig F2population[J].MolBiolRep,2010,37(1):467-471.
[5] SCHIAVO G,RIBANI A,UTZERI V J,et al.A genomic landscape of mitochondrial DNA insertions in the nuclear pig genome[J].JAnimSci,2016,94(S1):181.
[6] ZANG L,WANG Y D,SUN B X,et al.Identification of a 13 bp indel polymorphism in the 3′-UTR ofDGAT2 gene associated with backfat thickness and lean percentage in pigs[J].Gene,2016,576(2):729-733.
[7] XIAO Q,ZHANG Z,SUN H,et al.Genetic variation and genetic structure of five Chinese indigenous pig populations in Jiangsu Province revealed by sequencing data[J].AnimGenet,2017,48(5):596-599.
[8] XIAO Q,ZHANG Z,SUN H,et al.Pudong White pig:a unique genetic resource disclosed by sequencing data[J].Animal,2017,11(7):1117-1124.
[9] ZHANG Z,WANG Z,YANG Y,et al.Identification of pleiotropic genes and gene sets underlying growth and immunity traits:a case study on Meishan pigs[J].Animal,2016,10(4):550-557.
[10] CHEN Q,MA Y F,YANG Y M,et al.Genotyping by genome reducing and sequencing for outbred animals[J].PLoSOne,2013,8(7):e67500.
[11] YANG Y M,WANG Q S,CHEN Q,et al.A new genotype imputation method with tolerance to high missing rate and rare variants[J].PLoSOne,2014,9(6):e101025.
[12] WANG Z,CHEN Q,LIAO R,et al.Genome-wide genetic variation discovery in Chinese Taihu pig breeds using next generation sequencing[J].AnimGenet,2017,48(1):38-47.
[13] WANG Z,CHEN Q,YANG Y,et al.Genetic diversity and population structure of six Chinese indigenous pig breeds in the Taihu Lake region revealed by sequencing data[J].AnimGenet,2015,46(6):697-701.
[14] WANG Z,CHEN Q,YANG Y M,et al.A genome-wide scan for selection signatures in Yorkshire and Landrace pigs based on sequencing data[J].AnimGenet,2014,45(6):808-816.
[15] BROWNING B L,BROWNING S R.A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals[J].AmJHumanGenet,2009,84(2):210-223.
[16] HOWIE B N,DONNELLY P,MARCHINI J.A flexible and accurate genotype imputation method for the next generation of genome-wide association studies[J].PLoSGenet,2009,5(6):e1000529.
[17] FLICEK P,AHMED I,AMODE M R,et al.Ensembl 2013[J].NucleicAcidsRes,2013,41(D1):D48-D55.
[18] ZERBINO D R,WILDER S P,JOHNSON N,et al.The ensembl regulatory build[J].GenomeBiol,2015,16:56.
[19] ASHBURNER M,BALL C A,BLAKE J A,et al.Gene ontology:tool for the unification of biology[J].NatGenet,2000,25(1):25-29.
[20] KANEHISA M,GOTO S.KEGG:kyoto encyclopedia of genes and genomes[J].NucleicAcidsRes,2000,28(1):27-30.
[21] HUANG D W,SHERMAN B T,LEMPICKI R A.Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources[J].NatProtoc,2009,4(1):44-57.
[22] HUANG D W,SHERMAN B T,TAN Q,et al.DAVID bioinformatics resources:expanded annotation database and novel algorithms to better extract biology from large gene lists[J].NucleicAcidsRes,2007,35(W1):W169-W175.
[23] HU Z L,PARK C A,WU X L,et al.Animal QTLdb:an improved database tool for livestock animal QTL/association data dissemination in the post-genome era[J].NucleicAcidsRes,2013,41(D1):D871-D879.
[24] HU Z L,PARK C A,REECY J M.Developmental progress and current status of the Animal QTLdb[J].NucleicAcidsRes,2016,44(D1):D827-D833.
[25] YANG R,FANG S,WANG J,et al.Genome-wide analysis of structural variants reveals genetic differences in Chinese pigs[J].PLoSOne,2017,12(10):e0186721.
[26] DALL′OLIO S,FONTANESI L,TOGNAZZI L,et al.Genetic structure of candidate genes for litter size in Italian Large White pigs[J].VetResCommun,2010,34(S1):S203-S206.
[27] LIANG Q M,DENG H Y,SUN C W,et al.Negative regulation of IRF7 activation by activating transcription factor 4 suggests a cross-regulation between the IFN responses and the cellular integrated stress responses[J].JImmunol,2011,186(2):1001-1010.
[28] NING S,PAGANO J S,BARBER G N.IRF7:activation,regulation,modification and function[J].GenesImmun,2011,12(6):399-414.
[29] DIAMOND M S.IFIT1:A dual sensor and effector molecule that detects non-2′-O methylated viral RNA and inhibits its translation[J].CytokineGrowthFactorRev,2014,25(5):543-550.
