A First-Principles Study on the Vibrational and Electronic Properties of Zr-C MXenes∗
2018-05-14ChangYingWang王昌英YongLiangGuo郭永亮YuanYuanZhao赵媛媛GuangLiZeng曾广礼WeiZhang张伟CuiLanRen任翠兰HanHan韩晗andPingHuai怀平
Chang-Ying Wang(王昌英),Yong-Liang Guo(郭永亮),Yuan-Yuan Zhao(赵媛媛), Guang-Li Zeng(曾广礼),Wei Zhang(张伟),Cui-Lan Ren(任翠兰),Han Han(韩晗), and Ping Huai(怀平)
1Shanghai Institute of Applied Physics,Chinese Academy of Sciences,Shanghai 201800,China
2Department of Electronics and Communication Engineering,Henan Institute of Technology,Xinxiang 453003,China
3Department of Physics,School of Physics and Optoelectronic Engineering,Nanjing University of Information Science and Technology,Nanjing 210044,China
1 Introduction
Recently,a great achievement has been made in the synthesization of new Zr-C materials.[1]The new produced Zr3C2Tz(T=F,O,OH)is composed by the Zr3C2block and the functionalized group.It belongs to a new class of two-dimensional(2D)materials,MXene(Mn+1XnTx),which is a type of 2D transitional metal carbide,nitride and carbonitride and can be prepared from layered nanolaminates by selective etching method.[1−2]The terminal groups T in MXene are usually oxygen(O),hydroxyl(OH)or fl uorine(F).The currently researching hotspot of material science,MAX phase,is one kind of layered nanolaminates.[3−5]Taking Zr-C MXene as an example,its precursors can be Zrn+1AlCn(n=1,2)(MAX phase),(n=2,3)layered nanolaminates.[6]
In the past decades,more and more 2D materials are being investigated and synthesized for their excellent electronic properties,mechanical properties,optical properties and so on.[7−9]As the new discovered 2D materials,MXenes are currently expected to have numerous potential applications such as electronic devices[10−12]and electrode of Li ion batteries.[13−14]As the relatively early produced MXenes,2D Ti-C and Mo-C materials have been explored by many works within the framework of density functional theory(DFT).However the knowledge about Zr-C MXenes is scarce due to the late preparation of 2D Zr-based materials.Considering the fact that DFT has gained substantial achievements in material field,a firstprinciples investigations on the 2D ZrnCn−1(n=2,3,4)and their functionalized MXenes have been done in this work.[15−21]The geometry,phonon spectra,and electronic density of states(DOS)are explored to have a better understanding of the experimentally synthesized and theoretically potential Zr-C MXenes.
The rest of this work is organized as follows.The computational methods are described in Sec.2.The discussions on the structural properties,vibrational properties,and electronic properties of ZrnCn−1(n=2,3 and 4)and their functionalized MXenes are given in Sec.3.The conclusion part is presented in Sec.4.
2 Computational Methods
All calculations in this work were performed with the VASP (Vienna Ab-initio Simulation Package)code[22]with projector-augmented plane wave(PAW)potentials[23]and Perdew-Burke-Ernzerhof (PBE)exchange-correlation functional.[24]The wave functions were expanded in a plane-wave basis set with an energy cuto ffof 500 eV.A Γ-centeredk-point sampling scheme over the Brillouin zone was used for all the calculations.2D bare and functionalized MXenes were investigated with the vacuum space being larger than 17˚A to avoid any interaction of the MXene sheet with its periodically repeated images along thecaxis.A 18×18×2k-point mesh was used to optimize the structure of these 2D system.To check the dynamical stability of these 2D system,we calculated the phonon dispersion by using a supercell approach[25]as implemented in the PHONOPY code.[26]In these phonon calculations,a 4×4×1 supercell and 3×3×1k-point mesh were used.The symmetry nonequivalent Zr,Al and C atoms were displaced from their equilibrium positions by an amplitude of 0.02˚A and the atomic forces induced by these small displacements were calculated within VASP.In the calculation of the DOS for pristine and chemical functionalized MXenes,a 42×42×3k-point mesh was used.
3 Results and Discussions
3.1 Structural Properties
MXene can be prepared from layered nanolaminates by selective etching method.Theoretically there exist three types of Zr-C MXenes ZrnCn−1(n=2,3 and 4).They can be obtained from the Zr-Al-C nanolaminates(Zr2AlC(211),Zr3AlC2(312),Zr2Al3C4(234),Zr3Al3C5(335),Zr4Al3C6(436),Zr2Al4C5(245)and Zr3Al4C6(346)).The simulated Zr2C,Zr3C2and Zr4C3MXene structures as well as their possible precursors in Zr-Al-C system are depicted in Figs.1(a)–1(c).Generally,the pristine Zr-C MXenes are stabilized by the O,F,and OH functionalization during the etching process,thus generating nine kinds of functionalized MXenes.The top view of Zr2C is presented in Fig.1(d)and the possible adsorption sites(A,B and C)of functionalization atoms are also indicated in Fig.1(d).The chemical functionalization of the investigated MXenes have four possible con figurations:Con fi.1 has two functional groups adsorbed on the top of Zr atoms(site C),Con fi.2 has two functional groups located on the top of site B,Con fi.4 locates two functional groups on the top of site A and Con fi.3 is a combination of Con fi.2 and Con fi.4,in which one functional group locates on the top of site A and the other one on the top of site B.We compare the total energies of the four chemically adsorbed con figurations in Table 1 and find Con fi.2 has the lowest energy for all functionalized MXenes.This indicates that all functionalized species preferably adsorb on the top of site B.The side views of the most stable F,O,and OH functionalization structures of Zr2C are shown in Figs.1(e)–1(g),respectively.
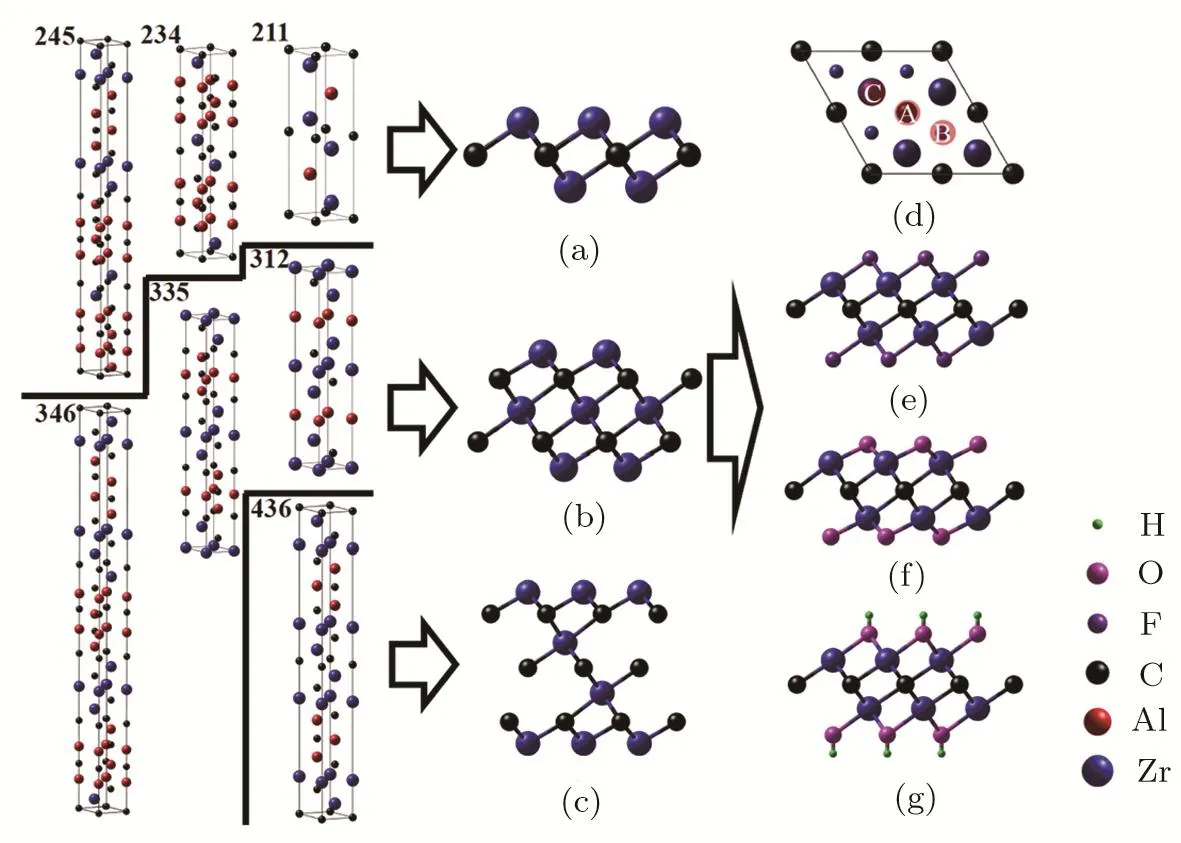
Fig.1 (Color online)Schematic illustrations of pristine and functionalized Zr-C MXenes.Side view of pristine Zr-C MXenes as well as their possible precursors in Zr-Al-C system:(a)Zr2C,(b)Zr3C2and(c)Zr4C3.(d)Top view of Zr2C.The large blue balls are the Zr atoms at the first layer and the small ones are those at the second layer.The symbol A represents the hollow site under which a carbon atom exists,B represents the hollow site under which there is no carbon atom,C is the top site of Zr atom.The rest three graphs are side views of(e)F,(f)O,and(g)OH functionalized Zr2C.The blue,red,black,purple,magenta and green balls indicate the Zr,Al,C,F,O and H atoms,respectively.
The optimized lattice parameters of bare ZrnCn−1(n=2,3,4)and their corresponding F,O and OH functionalized MXenes are shown in Table 2.The lattice parameters of functionalized Zr2C are all elongated with respect to the bare Zr2C MXene.The OH-terminated ZrnCn−1(n=2,3,4)MXene has a larger lattice parameter than its F and O-terminated counterparts.The Zr3C2O2and Zr4C3O2have the smallest lattice parameter value among Zr3C2and Zr4C3series,respectively.The binding energy of the functionalized group in ZrnCn−1T2(n=2,3,4)(T=F,O,OH)is calculated according to the following equation:[27]

where theandE(T)are the energies ofand the isolated functionalized group T,respectively.The binding energy can be used to predict the stabilities of the functionalizedMXenes.Our calculated binding energy of functionalizedMXenes are listed in Table 2.They are all large negative values,implying that the bareMXenes tend to be chemically decorated by the oxidizing group.On account of the computed binding energy,the stabilities of the functionalized Zr-C MXenes increase in the sequence of
3.2 Vibrational Properties
The stabilities of the bare and functionalized Zr-C MX-enes are explored by the calculation of their phonon spectra which are depicted in Fig.2.At the Γ point,the bare,and functionalized ZrnCn−1all have a quadratic fl exural acoustic phonon branch,similar to the other 2D materials,such as graphene,silicene,and phosphorene.[28−29]There exist no imaginary frequencies in all bare MXenes(Zr2C,Zr3C2and Zr4C3),indicating their dynamical stability.The imaginary frequency does not exist in the phonon dispersion of F,O,and OH functionalized Zr2C MXenes.The dynamical stability of these compounds indicates their possible existence,which is consistent with the previously reported results.[29]Moreover,the O-terminated Zr3C2and Zr4C3have no negative phonon modes,while the F and OH-terminated counterparts have small imaginary acoustic phonon branches around Γ point,which is consistent with the stabilities predicted from the binding energy of functionalized group.The imaginary frequency indicates the dynamical instability of F and OH-terminated Zr3C2and Zr4C3.However Zr3C2Tz(T=F,O,OH)MXenes have already been experimentally synthesized.[1]In consideration of the relatively small values of the calculated imaginary frequency,the unavoidable strain and temperature under experimental conditions might conquer the dynamical instability.

Table 1 Total energies(eV)of con figurations 1-4 for F,O and OH functionalized Zr2C,Zr3C2,and Zr4C3 systems.The most stable functionalization structure is labeled in bold.

Table 2 Optimized lattice parameters(˚A)and calculated binding energy(eV)of functionalized group in bare ZrnCn−1(n=2,3,4),as well as the most stable F,O and OH functionalized con figuration.
The phonon partial DOS of Zr-C MXenes are also calculated and depicted in Fig.2.In bare ZrnCn−1(n=2,3,4),the high-frequency phonon branches are contributed by the C atoms and the low-frequency ones come from the Zr atoms.In ZrnCn−1F2(n=2,3,4),the phonon dispersion spectra between 300 cm−1and 400 cm−1are mainly attributed to the vibration of the functionalized F atoms.In ZrnCn−1O2(n=2,3,4),the terminal O induced phonon vibrational frequencies are higher than the ones contributed by Zr atoms and lower than the ones caused by the C atoms.And the vibrational peak of the O atoms locates in the region of 400 cm−1and 450 cm−1.In ZrnCn−1(OH)2(n=2,3,4),the phonon branches above 3000 cm−1are the result of the vibrations of H atoms.In all calculated phonon partial DOS,the positions of the vibrational peaks have a close relationship with the atom species.In summary,the heavy atoms correspond to the low-frequency peaks,while the light atoms correspond to the high-frequency peaks.Except for the effect of the atomic mass,the interactions among atoms have also a signi ficant in fl uence on their vibrational frequencies.Take ZrnCn−1F2(n=2,3,4)as an example,the vibrational frequencies of F atoms are more close to the Zr atoms than the C atoms,even though its atomic mass is much closer to the C atoms.
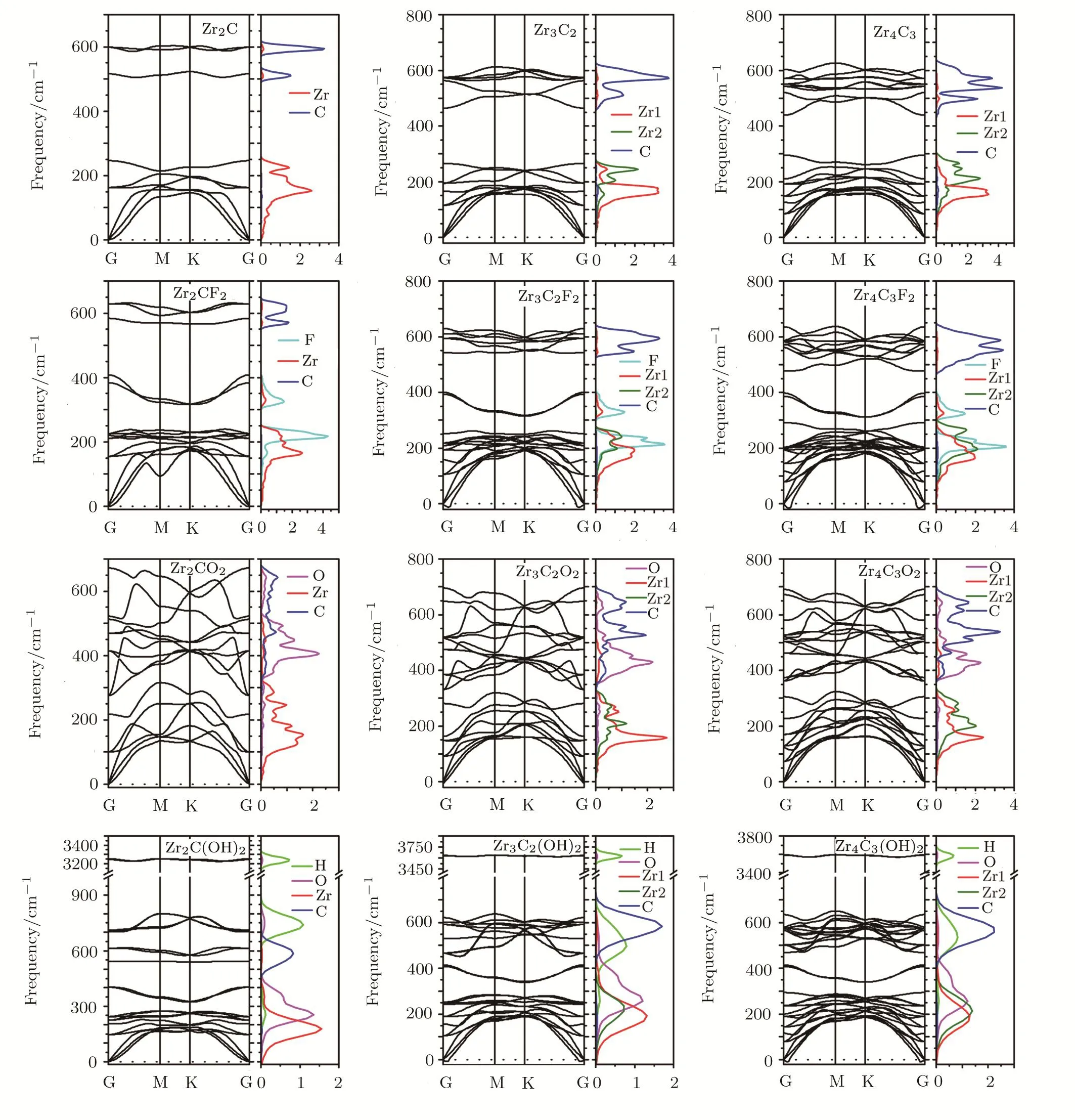
Fig.2(Color online)Phonon dispersion spectra and partial DOS of bare ZrnCn−1(n=2,3,4),as well as their most stable F,O,and OH functionalized con figuration.Zr represents the super ficial Zr layers,and Zr2 is the rest of the Zr layers.
3.3 Electronic Properties
The electronic properties of pristine and surface functionalized Zr-C MXenes have been calculated and presented in Fig.3–5.Pristine MXenes(Zr2C,Zr3C2,and Zr4C3)are all metallic with large DOS contributed by the Zr atom,especially the super ficial Zr layer,at the Fermi energy.The large Zr DOS peaks at the Fermi level might cause the instability of bare Zr-C MXenes,suggesting that the bare Zr-C MXenes tend to be functionalized by chemical groups.
When ZrnCn−1(n=2,3,and 4)MXenes are functionalized by F,O,and OH,new hybridization states appear and the DOS of MXenes at the Fermi level are reduced.The adsorbed F group hybridizes with the Zr atom at about−7.5 eV in all kinds of Zr-C MXenes and obtains one electron from the pristine ZrnCn−1(n=2,3,and 4).
The new hybridization state between hydrogen and oxygen atoms of the hydroxyl group locates around−9.5 eV in OH terminated Zr-C MXenes.Other additional bands in hydroxyl functionalized Zr-C MXenes are at about−6 eV and they are contributed by the interaction of the O atoms and the super ficial Zr atoms.The functionalized O atom gives rise to large DOS in ZrnCn−1O2(n=2,3,and 4)at about−4.4 eV and acquires two electrons from the pristine ZrnCn−1(n=2,3,and 4).The Fermi energy of the O terminated ZrnCn−1(n=2,3,and 4)MXenes shifts downward to the right edge of the hybridization state of C atom and Zr atom.In Zr2CO2,the Fermi level situates just at the band gap of the empty Ti band and the hybridized band of Zr,C and O.

Fig.3(Color online)Total and localized DOS of pristine and surface functionalized Zr2C MXenes.The vertical line represents the Fermi level.

Fig.4 (Color online)Total and localized DOS of pristine and surface functionalized Zr3C2MXenes.Zr1 represents the super ficial Zr layers,and Zr2 is the middle Zr layer.The vertical line represents the Fermi level.

Fig.5 (Color online)Total and localized DOS of pristine and surface functionalized Zr4C3MXenes.Zr1 represents the super ficial Zr layers,and Zr2 is the rest of the Zr layers.The vertical line represents the Fermi level.
In Zr3C2and Zr4C3,the super ficial Zr layers hybridize with the functionalized group in the low energy region,while the rest of the Zr atoms interact with the C atoms in the relatively high energy region.As thenvalue ofdecreases,the thicknesses of Zr slabs gradually decline,which lowers the DOS near the Fermi level.Especially for(ZrnCn−1)O2,Zr4C3O2is still a metal,Zr3C2O2is a semimetal,while Zr2CO2becomes a semiconductor with a band gap of 0.8 eV.
4 Conclusions
The fundamental properties of Zr-C MXenes are important to their potential applications.In this work,a systematic first-principles study on the structural,vibrational,and electronic properties of Zr-C MXenes have been conducted.The most stable con figurations for Zr-C MXene are the ones that the terminal groups F,O,and OH locate on the common hollow site of the super ficial Zr layer and its adjacent C layer.The stabilities of the functionalized Zr-C MXenes increase in the sequence ofaccording to the species of the terminal group.Among all the investigated MXenes,F and OH-terminated Zr3C2and Zr4C3have small imaginary acoustic phonon branches around Γ point while the others have no negative phonon modes.The pristine MXenes(Zr2C,Zr3C2and Zr4C3)are all metallic with large DOS contributed by the Zr atom at the Fermi energy.When functionalized by F,O and OH,new hybridization states appear and the DOS at the Fermi level are reduced.Especially for ZrnCn−1O2,Zr4C3O2is still a metal,Zr3C2O2is a semimetal,while Zr2CO2becomes a semiconductor.
We also thank the Supercomputing Center of Chinese Academy of Sciences(SCCAS)and the Shanghai Supercomputing Center for Computer Resources.
[1]J.Zhou,X.Zha,F.Y.Chen,et al.,Angew.Chem.Int.Ed.55(2016)5008.
[2]M.Naguib,M.Kurtoglu,V.Presser,et al.,Adv.Mater.23(2011)4248.
[3]Q.Huang,H.Han,R.Liu,et al.,Acta Mater.110(2016)1.
[4]H.Han,D.Wickramaratne,Q.Huang,et al.,RSC Adv.6(2016)84262.
[5]H.Wang,H.Han,G.Yin,et al.,Materials 10(2017)103.
[6]C.Y.Wang,H.Han,Y.Y.Zhao,et al.,J.Am.Ceram.Soc.101(2018)756.
[7]G.Fiori,F.Bonaccorso,G.Iannaccone,et al.,Nat.Nano.9(2014)768.
[8]F.Xia,H.Wang,D.Xiao,et al.,Nat.Photon.8(2014)899.
[9]D.Akinwande,N.Petrone,and J.Hone,Nat.Commun.5(2014)5678.
[10]M.Khazaei,M.Arai,T.Sasaki,et al.,Adv.Funct.Mater.23(2013)2185.
[11]A.N.Enyashin and A.L.Ivanovskii,J.Phys.Chem.C 117(2013)13637.
[12]M.Khazaei,A.Ranjbar,M.Ghorbani-Asl,et al.,Phys.Rev.B 93(2016)205125.
[13]J.Hu,B.Xu,C.Ouyang,et al.,RSC Adv.6(2016)27467.
[14]M.Naguib,J.Halim,J.Lu,et al.,J.Am.Chem.Soc.135(2013)15966.
[15]C.Y.Wang,H.Han,K.Shao,et al.,Chin.Phys.B 24(2015)097101.
[16]R.Yang,B.Tang,T.Gao,and B.Y.Ao,Commun.Theor.Phys.66(2016)447.
[17]Z.G.Wang,X.Y.Yang,S.J.Qin,and C.L.Wang,Commun.Theor.Phys.68(2017)125.
[18]N.Erum and M.A.Iqbal,Commun.Theor.Phys.66(2016)571.
[19]C.Y.Wang,H.Han,D.Wickramaratne,et al.,RSC Adv.7(2017)8421.
[20]K.Shao,H.Han,W.Zhang,et al.,Comp.Mater.Sci.137(2017)186.
[21]K.Shao,H.Han,W.Zhang,et al.,J.Nucl.Mater.490(2017)181.
[22]G.Kresse and J.Furthmüller,Phys.Rev.B 54(1996)11169.
[23]P.E.Blöchl,Phys.Rev.B 50(1994)17953.
[24]J.P.Perdew,K.Burke,and M.Ernzerhof,Phys.Rev.Lett.77(1996)3865.
[25]K.Parlinski,Z.Q.Li,and Y.Kawazoe,Phys.Rev.Lett.78(1997)4063.
[26]A.Togo,F.Oba,and I.Tanaka,Phys.Rev.B 78(2008)134106.
[27]T.Hu,Z.Li,M.Hu,et al.,J.Phys.Chem.C 121(2017)19254.
[28]G.Qin,Q.B.Yan,Z.Qin,et al.,Phys.Chem.Chem.Phys.17(2015)4854.
[29]Y.Uˇgur,ö.Ayberk,K.P.Nihan,et al.,Nanotechnology 27(2016)335702.
猜你喜欢
——张伟
杂志排行

Communications in Theoretical Physics的其它文章
- Cole-Hopf Transformation Based Lattice Boltzmann Model for One-dimensional Burgers’Equation∗
- Thermally Radiative Rotating Magneto-Nano fl uid Flow over an Exponential Sheet with Heat Generation and Viscous Dissipation:A Comparative Study
- Decoherence Effect and Beam Splitters for Production of Quasi-Ampli fied Entangled Quantum Optical Light
- Application of Connection in Molecular Dynamics
- Wilsonian Renormalization Group and the Lippmann-Schwinger Equation with a Multitude of Cuto ffParameters∗
- Higgs and Bottom Quarks Associated Production at High Energy Colliders in the Littlest Higgs Model with T-Parity∗