融合蛋白Trx-T4 DL可溶性表达、纯化与其生物活性应用
2018-04-26付大伟陈启蒙
付大伟,陈启蒙,徐 伟
(哈尔滨商业大学食品科学与工程重点实验室,黑龙江哈尔滨 150076)
T4 DNA连接酶(T4 DL)是基因工程技术中一种重要的工具酶,在体外可在ATP辅助下,催化粘端或平端双链DNA或RNA的连接[1-2],是DNA 复制过程中复制链的合成所必需的,广泛应用于体外 DNA连接、载体制备以及核酸分析等领域[3-5]。
传统的重组蛋白纯化方法是镍柱法,但存在操作程序繁琐、介质昂贵、纯化量少等问题,只适应于实验室规模的小量样品制备[6]。随着蛋白纯化技术的不断发展,近年来研发了磁珠法[7],因其操作简单、成本低廉、特异性强、纯化量大等,可应用于大规模生产样品等[8-9],但目前磁珠纯化法应用于重组蛋白纯化的研究较少。
目前,平末端PCR产物的连接,会出现平末端连接效率低,假阳性菌落多等现象,为解决这些问题,传统的方法是载体末端去磷酸化后再进行连接反应,但其存在操作繁琐,DNA 损失量较大,降低连接效率等问题,已成为急需解决的问题。
本文构建了含SUMO、IF2、GST、NusA、MsyB、Trx和MBP融合标签的重组表达载体,通过自诱导表达[10-11]及磁珠纯化法检测,筛选出高效表达的重组菌Transetta(DE3)(pNBEVII-T4 DL),并通过单因素实验、正交实验及方差分析法确定重组菌的最佳诱导培养条件。比较 MagNi 磁珠法与镍柱法对融合蛋白Trx-T4 DL的纯化效果,为磁珠法应用于重组蛋白的纯化提供理论依据。检测获得的融合蛋白Trx-T4 DL的连接活性,并与 T4 DNA焦磷酸激酶协同作用于低背景克隆载体的构建中,以提高连接效率和阳性率,为平末端PCR产物的连接提供一种新方法。
1 材料和方法
1.1 材料与仪器
Transetta(DE3)菌株、HiFi DNA Polymerase、Taq DNA Polymerase、DNA Marker和EcoR V 北京全式金生物技术有限公司;Trans I菌株和E.coliBL21(DE3)菌株 实验室保存;本实验所用质粒及其特性说明见表1。

表1 实验室所用质粒Table 1 Plasmids used in this work
限制性内切酶EcoR I、BamH I、Not I和Hind Ⅲ NEB公司;一步法克隆试剂盒 南京诺唯赞生物科技有限公司;异丙基硫化-β-半乳糖苷(IPTG) Sigma公司;氨苄青霉素(Ampr)、卡那霉素(Kana)、氯霉素(Cmr)三羟甲基氨基甲烷(Tris)、二硫苏糖醇(DTT)、Triton X-100、苯甲基磺酰氟(PMSF) Taraka公司;琼脂糖凝胶DNA回收试剂盒D2500-01、GenEluteTMPlasmid Miniprep Kit 美国Omega公司;Ni-NTA柱 Novagen公司;蛋白质marker Thermo公司;MagNi Protein Purification Kit、脱盐柱 北京诺贝奥生物技术有限公司。
T100 TM Thermal Cycler PCR仪 美国Bio-Rad公司;DYY-6C型电泳仪 北京六一仪器厂;H1650-W台式离心机 湖南湘仪实验室仪器开发有限公司;GDS8000凝胶成像系统 美国UVP公司;W201B恒温水浴锅 上海申胜生物技术有限公司;DW-86L388A立式超低温保存箱 青岛海尔特种电器有限公司;DHP-9162恒温培养箱、HZQ-311C落地恒温振荡器 上海恒一科学仪器有限公司;LDZX-50KB立式压力蒸汽灭菌器 上海申安医疗器械厂。
1.2 实验方法
1.2.1 T4 DL基因的扩增及克隆载体构建 根据T4 DL的基因在Genbank数据库中报道的序列(EU118292)及所需载体的基因序列来设计引物,在上、下游引物的5′端分别加入了BamH I和Not I酶切位点。
上游引物P1:5′-TGTATTTTCAGGGAGGATCC ATGATTCTTAAAATTCTGAACGAAATAGC-3′,下游引物P2:5′-GGTGGTGCTCGAGTGCGGCCGCTCA TAGACCAGTTACCTCATGAAAATC-3′(下划线代表酶切位点),引物送至上海生工生物有限公司进行合成。以提取的T4噬菌体(T4 bacteriophage)的基因组为模板,利用上述P1、P2引物进行PCR扩增。扩增产物经1% 琼脂糖凝胶电泳检测后,用DNA凝胶回收试剂盒进行纯化回收。通过一步法将纯化产物与经BamH I和Not I酶切后的pET-22b(+)表达载体连接,构建重组载体 pET-22b(+)-T4 DL,并直接转化到E.coliTrans I克隆感受态细胞中,利用T7和P2引物进行菌落 PCR检测阳性克隆,反应条件为:95 ℃,5 min;95 ℃,30 s;50 ℃,40 s;72 ℃,1 min;34个循环;72 ℃,5 min;终止反应,再利用BamH I和Not I双酶切鉴定阳性克隆,并将其送到上海生工生物有限公司进行序列测定。
1.2.2 融合蛋白的表达载体构建 按1.2.1中方法把测序成功的T4 DL基因与不同融合蛋白标签的表达载体通过一步法进行连接,构建融合蛋白表达载体,转化到E.coliTrans I感受态中,涂布于含Ampr的LB平板上,按1.2.1中方法鉴定阳性克隆。
1.2.3 自诱导表达的融合蛋白的可溶性表达
1.2.3.1 重组菌的自诱导表达 分别将检测成功的重组载体转化到E.coliTransetta(DE3)表达感受态细胞中,涂布于含Ampr及Cmr的LB固体培养基上,挑取单个菌落,接种于含Ampr和Cmr的1 mL LB 培养液中,37 ℃振荡培养过夜,再按1‰(V/V)的接种量接种于10 mL ZYP+5052自诱导培养基中[14-15],37 ℃,150 r/min诱导培养14 h,离心收集菌体,10% SDS-PAGE电泳分析蛋白表达情况。
1.2.3.2 融合蛋白的可溶性检测 取1.2.3.1中诱导表达的重组菌液1 mL,12000 r/min离心5 min,收集菌体,按MagNi Protein Purification Kit的说明书进行纯化。取沉淀、上清、穿透、洗脱液进行10% SDS-PAGE分析,选出表达量较高的菌株。
1.2.4 重组菌E.coliTransetta(DE3)(pNBEVII-T4 DL)培养条件单因素实验 选择影响发酵的4种培养条件:温度、接种量、pH、装瓶量进行单因素实验,每种实验重复3次,按1.2.4.5方法进行检测,其结果取无显著差异的数值的平均值,确定各因素的最佳条件[12]。
1.2.4.1 培养温度的筛选 接1‰(V/V)菌种于装有初始pH7的10 mL ZYM-5052 Ampr和Cmr培养基的 50 mL三角瓶中,16、25、30、37、42 ℃,150 r/min振荡培养16 h,筛选出最适温度。
1.2.4.2 接种量的筛选选择 不同接种量:1‰、5‰、10‰、15‰、20‰、30‰(V/V)接菌种于装有初始pH为7的10 mL ZYM-5052 Ampr和Cmr培养基的50 mL三角瓶中,37 ℃,150 r/min振荡培养16 h,筛选出最适接种量。
1.2.4.3 初始pH的筛选 接1‰(V/V)菌种于装有初始pH分别为5、6、7、8、9的10 mL ZYM-5052 Ampr和Cmr培养基的50 mL三角瓶中,37 ℃,150 r/min振荡培养16 h,筛选出最适接PH。
1.2.4.4 装瓶量的筛选 接1‰(V/V)菌种于装有初始pH为7的 25、50、75、100、150 mL ZYM-5052 Ampr和Cmr培养基的250 mL三角瓶中,37 ℃,150 r/min振荡培养16 h,筛选出最适瓶装量[12]。
1.2.4.5 分析方法 菌体密度的测定:取1.2.3中菌液稀释合适倍数,用分光光度计测定 600 nm 处的吸光值,其乘以稀释倍数即发酵液的菌体密度(OD600);融合蛋白Trx-T4 DL的可溶性检测:按1.2.3.2中方法检测,取样进行10% SDS-PAGE分析及Bandscan软件分析蛋白可溶性表达量;可溶性融合蛋白的产量测定:取上一步骤中的洗脱液用大分子脱盐试剂盒进行脱盐,用BCA法测定其中融合蛋白Trx-T4 DL浓度。
1.2.4.6 正交设计实验优化 培养条件采用L9(34)正交表进行培养条件优化实验,以温度、装瓶量、接种量及pH为研究对象(表2),在转速为150 r/min的摇床中振荡培养16 h,每个处理3次重复,以蛋白产量值分析4因素3水平对重组菌表达量的影响,以确定最佳的培养条件[12]。

表2 正交实验因素水平表Table 2 The orthogonal experiment factor level table
1.2.5 镍柱和 MagNi 磁珠纯化效果的比较
1.2.5.1 菌体破碎 将菌液按2‰(V/V)的接种量接种于装瓶量为50 mL/250 mL、pH7的100 mL含Ampr及 Cmr的ZYP+5052自诱导培养基中,30 ℃培养14 h,离心收集菌体,加入结合缓冲液,终浓度1 mg/mL的溶菌酶,10 mmol/L PMSF及终浓度为1%的Triton-X 100充分混匀,冰浴30 min,超声破碎[13],离心收集上清。
1.2.5.2 Ni-NTA亲和层析柱纯化 用Ni-NTA柱(Novagen)进行亲和层析纯化[14],对收集的上清进行10% SDS-PAGE分析。
1.2.5.3 MagNi磁珠纯化 按1.2.3.2中方法进行纯化,对收集的沉淀、上清、穿透、洗脱液进行10% SDS-PAGE分析。
1.2.6 融合蛋白Trx-T4 DL的浓度检测 包埋法对1.2.5.3中纯化的融合蛋白Trx-T4 DL进行浓缩,经脱盐柱脱盐以除去咪唑,用BCA法测定其浓度,加入贮存缓冲液,-20 ℃保存。
1.2.7 融合蛋白Trx-T4 DL的活性检测 依照周李华等[15]采取的方法进行活性测定。按表3配置连接反应体系,反应体系中以ddH2O代替T4 DL设为空白对照,以T4 DL作对照。轻轻混匀,16 ℃水浴30 min,反应结束后65 ℃水浴10 min,置于冰上2 min。电泳检测连接情况,按连接情况加入酶的最小体积乘以稀释倍数计算酶活力。
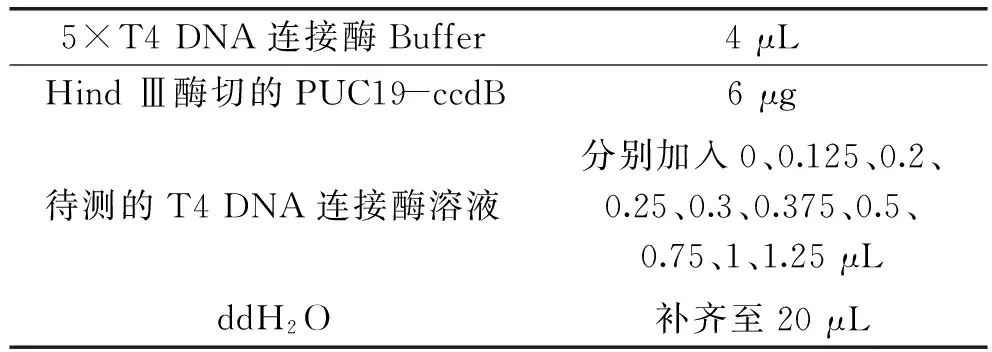
表3 酶活力测定的反应体系Table 3 The reaction system of enzyme activity detection
1.2.8 融合蛋白Trx-T4 DL在低背景重组载体构建中的应用 以EcoR V酶切的PUC19-ccdB载体和PCR扩增的平末端TEV片段为底物,按表4配置连接反应体系,25 ℃反应 15 min,置冰上 2 min,以PUC19-ccdB质粒及不加T4 DL为空白对照,以传统连接方法做对照,热激转化到E.coliTrans I克隆感受态中细胞中,涂布于含Ampr和Kana的SOB固体培养基中,37 ℃培养过夜。对过夜培养出的菌落进行PCR检测。按1.2.1中方法鉴定阳性克隆,并计算阳性率。

表4 连接反应体系Table 4 The ligation reaction system
1.3 数据统计分析
实验中每个处理重复3次,采用正交助手软件进行数据的方差分析,应用Excel 2013软件作表,应用10% SDS-PAGE及Bandscan软件分析蛋白表达量,应用Adobe Photoshop CC 2014软件作图。
2 结果与分析
2.1 T4DL基因扩增及克隆的检测
2.1.1 T4 DL基因的扩增 以T4噬菌体基因组为模板经PCR扩增,1%琼脂糖凝胶电泳显示在约 1464 bp处有一条清晰带(图1),与预期目的片段大小一致。
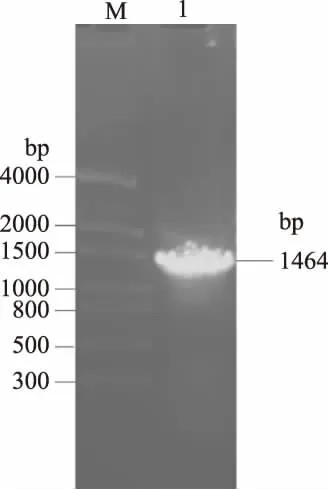
图1 PCR扩增T4 DL基因Fig.1 PCR Amprlification of T4 DL gene注:M:DNA marker;1:T4 DL基因PCR产物。
2.1.2 菌落PCR 扩增出的目的条带经一步法连接到pET-22b(+)表达载体上,转化到E.coliTrans I中,涂布于含Ampr的LB固体培养基,37 ℃培养过夜,挑取7个阳性菌落进行菌落PCR检测,1%琼脂糖凝胶电泳显示在约1600 bp处都有一清晰带条,与预期目的片段大小一致(图2),以空载体为阴性对照无目的条带,说明目的条带成功插入pET-22b(+)表达载体中。
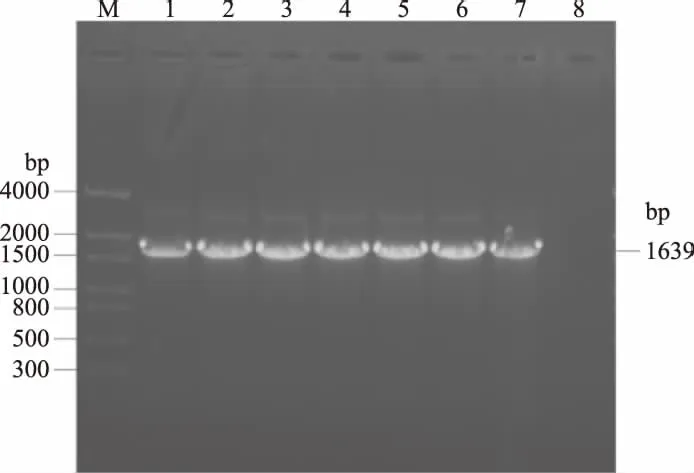
图2 菌落PCR鉴定pET-22b(+)-T4 DL重组质粒Fig.2 Identification of recombinant plasmidpET-22b(+)-T4 DL by PCR注:M:DNA marker;1~7:重组菌株;8:空载体。
2.1.3 酶切及测序鉴定 取菌落PCR检测成功的菌株进行培养,按GenEluteTMPlasmid Miniprep Kit进行质粒的提取,以重组质粒为阴性对照,对重组质粒分别进行BamH I和Not I酶切。1%琼脂糖凝胶电泳检测,可看到约1464 bp和约5370 bp两条条带(图3),与预期大小基本一致。利用NCBI中的BLAST程序,将测序的结果与GenBank中的基因序列进行比对,相似性达 98%,目的片段成功插入到载体中。

图3 酶切鉴定pET-22b(+)-T4 DL重组质粒Fig.3 Identification of recombinant plasmidpET-22b(+)-T4 DL by double enzyme digestion.注:M:DNA marker;1:pET-22b(+)-T4 DL重组质粒;2:BamH I和Not I酶切pET-22b(+)-T4 DL重组质粒。
2.2 重组表达载体的构建
以成功构建的重组质粒pET-22b(+)-T4 DL为模板,扩增出的T4 DL基因经一步法分别连接BamH I和Not I酶切后的pNBE Ⅰ~Ⅷ表达载体(图4),转化到E.coliTrans I中,涂布于含Ampr的LB固体培养基,37 ℃培养过夜,每种重组菌挑取4个阳性菌落进行菌落PCR检测(图5),1% 琼脂糖凝胶电泳显示,pNBE I~Ⅷ中有目的条带出现,其约 1655 bp,与预期大小基本一致,说明重组表达载体构建成功。

图4 酶切鉴定重组表达载体Fig.4 Identification of the recombinant plasmid by restriction enzyme digestion 注:M:DNA marker;1:PCR 扩增的T4 DL基因;2~9:BamH I和Not I酶切的重组质粒pNBE I~Ⅷ。

图5 菌落PCR鉴定重组表达载体Fig.5 Identification of the recombinant plasmid by PCR注:M:DNA marker;1~4:重组表达载体pNBE I-T4DL;5~8:重组表达载体pNBE II-T4DL;9~12:重组表达载体pNBE IIII-T4DL;13~16:重组表达载体pNBE IV-T4DL;17~20:重组表达载体pNBE V-T4DL;21~24:重组表达载体pNBE VI-T4DL;25~28:重组表达载体pNBE VII-T4DL;29~32:重组表达载体pNBE Ⅷ-T4DL。
2.3 重组菌的自诱导表达
选取检测成功的菌株,按GenEluteTMPlasmid Miniprep Kit进行质粒提取,转化到E.coliTransetta(DE3)表达感受态细胞中,筛选菌株进行自诱导培养,收集菌体,10% SDS-PAGE电泳显示(图6),重组菌Transetta(DE3)/pNBE II-T4DL、Transetta(DE3)/pNBE III-T4DL、Transetta(DE3)/pNBE IV-T4DL、Transetta(DE3)/pNBE VI-T4DL、Transetta(DE3)/pNBE VII-T4DL表达的与预期蛋白大小相符的融合蛋白,大小分别为77.41、97.81、112.81、89.81、75.11 kDa。
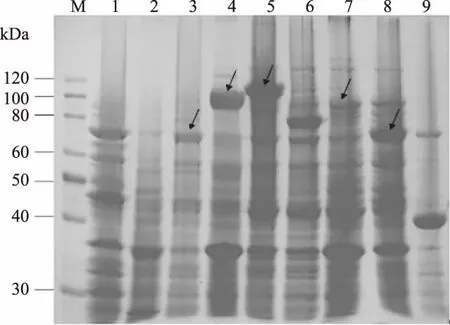
图6 T4 DL诱导表达的10% SDS-PAGE分析Fig.6 The 10% SDS-PAGE profile of T4 DL induced expression 注:M.蛋白质 marker;1:未诱导菌体;2~9:分别为自诱导的表达载体为pNBE I~Ⅷ的重组菌。
2.4 融合蛋白的可溶性检测
对1.2.3.1 中收集的菌体进行磁珠纯化检测,以确定各重组菌表达可溶性T4 DL的情况,由10% SDS-PAGE电泳显示(图7),重组菌Transetta(DE3)/pNBE II-T4 DL、Transetta(DE3)/pNBE III-T4 DL、Transetta(DE3)/pNBE VII-T4DL经纯化检测后在洗脱液中有目的蛋白条带,说明其表达可溶性融合蛋白,而纯化后条带较为单一的是重组菌Transetta(DE3)/pNBE VII-T4DL,表达的可溶性融合蛋白Trx-T4 DL较多,大小约70 kDa,与预期的分子质量大小相符。随后对该重组菌进行培养条件的优化,以增加其表达量。

图7 T4 DL可溶性表达的10% SDS-PAGE分析Fig.7 The 10% SDS-PAGE profile of T4 DL expressed in a soluble form注:M:蛋白 marker;1,5,9,13,17,21,25,29:分别为含表达载体pNBE Ⅰ、Ⅱ、Ⅲ、Ⅳ、Ⅴ、Ⅵ、Ⅶ、Ⅷ的重组菌破碎后沉淀;2,6,10,14,18,22,26,30:分别为含表达载体pNBE Ⅰ、Ⅱ、Ⅲ、Ⅳ、Ⅴ、Ⅵ、Ⅶ、Ⅷ的重组菌破碎后上清;3,7,11,15,19,23,27,31:分别为含表达载体pNBE Ⅰ、Ⅱ、Ⅲ、Ⅳ、Ⅴ、Ⅵ、Ⅶ、Ⅷ重组菌破碎后纯化的穿透;4,8,12,16,20,24,28,32:分别为含表达载体pNBE Ⅰ、Ⅱ、Ⅲ、Ⅳ、Ⅴ、Ⅵ、Ⅶ、Ⅷ重组菌破碎后纯化的洗脱。
2.5 培养条件的优化
2.5.1 单因素实验优化培养条件 本实验选择对菌体培养影响较大的4个方面:温度、接种量、pH、装瓶量,分别对其进行单因素实验,各因素取值及结果见表5。由表5可知,温度对菌体浓度、蛋白可溶性表达量及蛋白产量均有较大影响,对菌体浓度而言,温度越高菌体密度越大,说明在一定时间及温度内,菌体生长速度越快,整体呈上升趋势;对蛋白可溶性表达量而言,随着温度升高蛋白可溶性表达量和蛋白产量均先升高后降低,温度为30 ℃时可溶性表达量最高,为65.36%,蛋白产量为646.470 mg/L,其原因是温度越高菌体生长较快,菌体生长及表达越多,目的蛋白表达速度也过快,蛋白错误折叠率随之增加,形成包涵体增多,进而可溶性蛋白减少,进而目的蛋白产量减少,但温度过低菌体生长及表达速度都过慢,可溶性蛋白减少,说明目的蛋白产量受蛋白可溶性表达量影响较大。

表5 培养条件单因素实验设计及结果Table 5 Single factor experiments design and results
接种量对菌体浓度、蛋白可溶性表达量及蛋白产量均有影响,对菌体浓度而言,接种量越高菌体生长速度越快,菌体密度值越大;对可溶性表达量而言,接种量越多蛋白可溶性表达量越多,最适温度5‰,对应的蛋白可溶性表达量为56.28%,随后可溶性表达量逐渐降低,其原因是接种量过多菌体生长及表达速度过快,蛋白错误折叠率增加,形成包涵体增多,进而可溶性蛋白减少;对蛋白产量而言,其最适温度也为5‰,对应的蛋白产量为548.359 mg/L,其结果与蛋白可溶性表达量的一致,说明目的蛋白产量受蛋白可溶性表达量影响较大。
装瓶量对菌体浓度、蛋白可溶性表达量及蛋白产量均有影响。最适瓶装量为50 mL/250 mL,此时的菌体密度为3.58,蛋白可溶性表达量为56.55%,蛋白产量为614.012 mg/L,其原因是瓶装量越多培养基多,菌体浓度增加,瓶装量越多菌体生长及表达越快,蛋白可溶性表达量越多,但瓶装量过多溶氧量减少,严重影响菌体生长速度,会导致目的蛋白错误折叠率增加,形成的包涵体增加,蛋白可溶性表达量减少,菌体浓度降低,说明目的蛋白产量受菌体浓度及蛋白可溶性表达量影响。
初始pH对菌体浓度、蛋白可溶性表达量及蛋白产量均有影响。对菌体浓度而言,最适初始pH为6,其菌体密度为2.75,说明培养环境的pH对菌体的生长起到关键性的作用;对蛋白可溶性表达量而言,最适初始pH为7,对应的蛋白可溶性表达量为53.01%,其原因是过酸或过减都影响蛋白稳定性,使目的蛋白聚沉形成包涵体;对蛋白产量而言,最适初始pH为7,对应的蛋白产量为544.247 mg/L,与蛋白可溶性表达量一致,说明目的蛋白产量受蛋白可溶性表达量影响较大。
通过上述单因素实验可知,菌体密度和蛋白可溶性表达量共同影响着蛋白的最终产量,且优化的最终目的是提高蛋白产量,所以后续实验以蛋白产量为指标,省略菌体密度及蛋白可溶性表达量的测定。
2.5.2 正交设计实验优化培养条件 以温度、接种量、装瓶量、pH为研究对象,以蛋白产量为指标,进行四因素三水平的正交实验,确定培养条件的最佳组合,实验设计及结果分析见表6和表7。

表6 L9(34)正交设计表及结果分析Table 6 The L9(34)orthogonal design table and the analysis of results
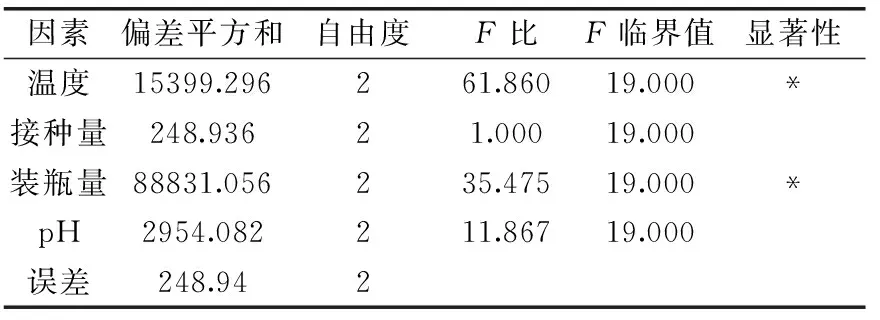
表7 正交实验方差分析Table 7 Variance analysis of orthogonal test
从直观分析(表6)可知,RA>RC>RD>RB,表明4 种培养条件对蛋白产量的影响由大到小依次为:温度、装瓶量、pH、接种量。从方差分析(表7)可知,影响目的蛋白产量的各因素显著性次序为温度>装瓶量>pH>接种量,其中温度和瓶装量均有显著性影响。通过正交实验获得发酵培养条件的最佳组合为A2B2C3D2,但在正交设计表中并没有该组合的实验,为此,我们按此组合补充验证实验,在此条件下,目的蛋白产量达744.023 mg/L,高于正交试验组合各组结果。综合以上实验结果,确定最佳诱导培养条件:温度30 ℃、装瓶量50 mL/250 mL、pH7、接种量2‰。
2.6 镍柱和MagNi磁珠纯化效果的比较
2.6.1 Ni-NTA亲和层析柱纯化 对重组菌Transetta(DE3)/pNBE VII-T4DL进行破碎,取5 mL裂解后上清液进行亲和层析纯化,10% SDS-PAGE显示(图9),纯化后的洗脱液有一条清晰条带,其大小为75.11 kDa,与预期相同,但并不是单一条带,纯化程度不高,并且穿透及漂洗液中都含有融合蛋白,说明结合不完全,最终获得的融合蛋白量少。
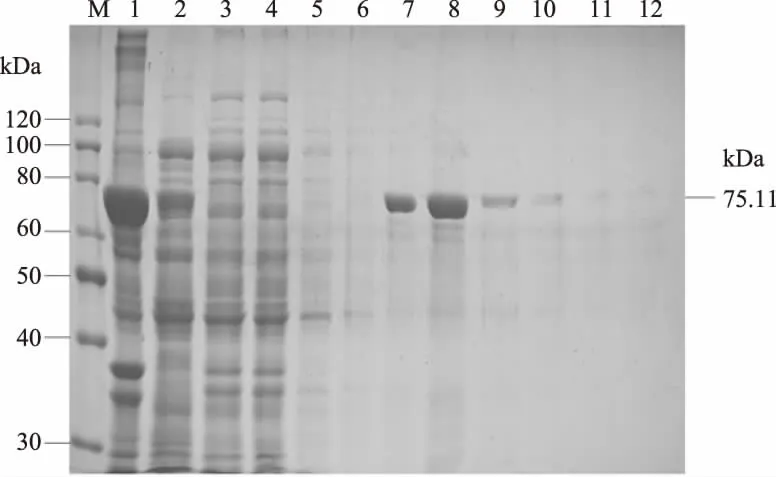
图9 Ni-NTA柱纯化T4 DL的10% SDS-PAGE分析Fig.9 The 10% SDS-PAGE analysis result of T4 DL purified by Ni-NTA column注:M:蛋白质 marker;1:纯化前重组菌裂解后的沉淀;2:纯化前重组菌裂解后的上清;3:穿透液;4~6.洗涤液;7~12:第1次到第6次分批洗脱收集的T4 DL。
2.6.2 MagNi磁珠纯化 取5 mL裂解后上清液进行亲和层析纯化,由10% SDS-PAGE显示(图10),虽穿透中有少许T4 DL,但漂洗中融合蛋白很少,说明结合较完全,洗脱液仅有一条清晰条带,其大小为75.11 kDa,与预期相同,纯化程度较高,纯化量高,所以采用MagNi磁珠纯化T4 DL效果较好。
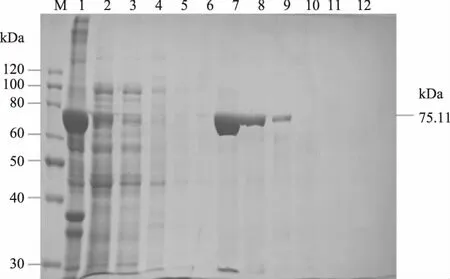
图10 MagNi磁珠纯化T4 DL的10% SDS-PAGE分析Fig.10 The 10% SDS-PAGE analysis result of T4 DL purified by MagNi magnetic beads注:M:预染蛋白质 marker;1:纯化前重组菌裂解后的沉淀;2:纯化前重组菌裂解后的上清;3:穿透液;4~6.洗涤液;7~12:第1次到第 6次分批洗脱收集的T4 DL。
2.7 融合蛋白Trx-T4 DL的浓度测定
分别对2.7中镍柱和MagNi磁珠纯化的融合蛋白Trx-T4 DL进行浓缩、脱盐,采用BCA法测定其浓度分别为559.29、1700.462 mg/L。
2.8 T4 DNA连接酶酶活力测定
本文以A公司购买的T4 DL作对照,1% 琼脂糖凝胶电泳显示(图11),当A公司购买的T4 DL加入量超过 0.75 μL时,955 bp和3944 bp片段消失,经Hind Ⅲ 酶切底物成功连接,而获得的T4 DL加入超过 0.3 μL时,经 Hind Ⅲ 酶切底物成功连接,以A 公司购买的T4 DL作对照,根据稀释倍数可计算出酶活力约为500 U/μL。

图11 T4 DL酶活力测定的10% SDS-PAGE分析Fig.11 The 10% SDS-PAGE analysis result of the enzyme activity determination of T4 DL注:M:DNA marker,1~10:本实验获得的T4 DL各浓度梯度连接情况;11~20:A公司的T4 DL各浓度梯度连接情况。
2.9 融合蛋白T4 DL在低背景重组载体构建中的应用
由表8可知,含PUC19-ccdB质粒的菌体不生长,说明ccdB致死基因具有抑制宿主细胞生长的作用,不加T4 DL的反应体系没有菌体生长,说明载体酶切较为完全或自连的空载体会抑制宿主细胞生长,说明PUC19-ccdB载体可提高阳性重组率,从而加速阳性克隆筛选进程[16-18]。与传统连接方法相比,阳性菌落数多,阳性率可达96.87%,说明该方法连接效率更高。

表8 不同连接方法的比较Table 8 Comparison of different ligation methods
3 结论与讨论
本文构建了含SUMO、IF2、GST、NusA、MsyB、Trx和 MBP融合标签的重组表达载体,通过磁珠纯化法检测可溶性表达情况,筛选出高效表达的重组菌Transetta(DE3)(pNBEVII-T4 DL),并采用单因素实验和正交设计实验来优化诱导培养条件,通过方差分析法确定最佳诱导培养条件:温度30 ℃、装瓶量50 mL/250 mL、pH7、接种量2‰,比优化前的蛋白产量增加了约100 mg/L。利用MagNi 磁珠纯化获得大量高纯度融合蛋白Trx-T4 DL,经BCA法检测其浓度为1700.462 mg/L,与镍柱法相比,其浓度提高了约3倍。依照周李华等[15]采取的方法进行活性测定,获得的融合蛋白Trx-T4 DL的酶活约为500 U/μL。分别利用本实验的连接方法与传统连接方法进行低背景克隆载体构建,阳性克隆率高达96.87%,证明该方法具有高效性。
本文为增加可溶性T4 DL的产量分别通过增加融合标签、选择Transetta(DE3)作为宿主菌和优化诱导培养条件,但由于蛋白质的种类繁多,理化性质各异,目前没有一个可以提高可溶性表达的通用方法,所以最佳表达可溶性T4 DL的方法可进一步深入研究。通过上述可知,高温与瓶装量对重组菌体Transetta(DE3)(pNBEVII-T4 DL)的可溶性表达影响较为显著,因此,我们可降低适当培养温度,减少瓶装量以增加蛋白产量。目的蛋白只有正确折叠能可溶性表达,这也与溶氧量有关,在后续实验中可以讨论溶氧量与目的蛋白产量的相关性,可以通过控制通氧速率来恒定溶氧量,以提高目的蛋白产量。
[1]Murray N E,Bruce S A,Murray K. Molecular cloning of the DNA ligase gene from bacteriophage T4 II Amprlification and preparation of the gene product[J]. Journal of Molecular Biology,1979,132:493-505.
[2]Zhao B,Tong Z,Zhao G,et al. Effects of 2′-O-methyl nucleotide on ligation capability of T4 DNA ligase[J]. Acta Biochim Biophys Sin,2014,46:727-737.
[3]Onda M,Yamaguchi J,Hanada K,et al. Role of DNA ligase in the illegitimate recombination that generates lambdabio-transducing phages inEscherichiacoli[J]. Genetics,2001,158(1):29-39.
[4]Lohman G,Chen L X,Evans T C. Kinetics characterization of single strand break ligation in duplex DNA by T4 DNA ligase[J].Journal of Biology Chemistry,2011,286:44187-44196.
[5]韩来闯,马闪闪,刘亚娟,等. 构建重组质粒的二步 PCR方法[J]. 河南科学,2015,33(8):1321-1325.
[6]Kalim M,Jie C,Wang S,et al. Construction of high level prokaryotic expression and purification system of PD-L1 extracellular domain by usingEscherichiacoli,host cell machinery[J]. Immunology Letters,2017,190:34-41.
[7]Dammicco S,Goux M,Lemair C,et al. Regiospecific radiolabelling of nanofitin on Ni magnetic beads with[18F]FBEM andinvivoPET studies[J]. Nuclear Medicine and Biology,2017,51:33-39.
[8]Vereshchagina T A,Fedorchak M A,Sharonova O M,et al. Ni(2+)-zeolite/ferrosphere and Ni(2+)-silica/ferrosphere beads for magnetic affinity separation of histidine-tagged proteins[J]. Dalton Transactions,2015,45(4):1582-92.
[9]Rosenbaum A,Bleck E,Schneider M,et al. Assessment of direct versus indirect magnetic bead-based T-cell isolation procedures followed by magnetic bead-based DNA isolation[J]. Lupus Science & Medicine,2016,3(1):e000167.
[10]Nie Y,Yan W,Xu Y,et al. High-level expression ofBacillusnaganoensis pullulanase from recombinantEscherichiacoliwith auto-induction:effect of lac operator[J]. Plos One,2013,8(10):e78416.
[11]马闪闪,韩来闯,刘亚娟,等. CBM2蛋白的高密度自诱导[J]. 中国农学通报,2015,31(18):140-145.
[12]顾娟,劳勋,金明飞,等. 人胰高血糖素样肽-1突变体融合蛋白在大肠杆菌中的自诱导表达优化[J]. 微生物学通报,2010,37(5):726-731.
[13]Joshi H,Jain V. Novel method to rapidly and efficiently lyseEscherichiacolifor the isolation of recombinant protein[J]. Analytical Biochemistry,2017,528:1-6.
[14]Alves N J,Turner K B,Divito K A,et al. Affinity purification of bacterial outer membrane vesicles(OMVs)utilizing a His-tag mutant[J]. Research in Microbiology,2017,168(2):139-146.
[15]周李华,叶德萍,王智,等. T4 DNA连接酶酶活力测定方法[J].中国测试,2014,40(2):64-66.
[16]Zhang Q,Yan Z,Xu Y,et al. Characterization of inducible ccdB gene as a counterselectable marker inEscherichiacolirecombineering[J]. Current Microbiology,2017,74(8):961-964.
[17]Baliga C,Varadarajan R,Aghera N. The homodimericE.colitoxin CcdB(Controller of Cell Division or Death B protein)folds via parallel pathways[J]. Biochemistry,2016,55(43):6019-6031.
[18]耿晓姗,刘秦,党会杰,等. 利用毒素蛋白基因ccdB构建高效低背景T-载体[J]. 热带生物学报,2016,7(2):232-236.
