Mayaro virus infection, the next epidemic wave after Zika? Evolutionary and structural analysis
2018-04-26EleonoraCellaMartaGiovanettiTeresaMilanoMartaFogolariFrancescoGarilliIvailoAlexievRiccardoBazzardiMarcoSalemiLuizCarlosJuniorAlcantaraSilviaAngelettiStefanoPascarellaMassimoCiccozzi
Eleonora Cella, Marta Giovanetti, Teresa Milano, Marta Fogolari, Francesco Garilli, Ivailo Alexiev, Riccardo Bazzardi, Marco Salemi, Luiz Carlos Junior Alcantara, Silvia Angeletti✉,Stefano Pascarella, Massimo Ciccozzi✉
1Public Health and Infectious Diseases, Sapienza University, Rome, Italy
2Unit of Clinical Laboratory Science, University Campus Bio-Medico of Rome, Italy
3Fundação Oswaldo Cruz, Salvador, Bahia, Brazil
4University of Rome “Tor Vergata”, Rome, Italy
5Dipartimento di Scienze Biochimiche "A. Rossi Fanelli", Università La Sapienza, 00185 Roma, Italy
6Department of Medicine, University Campus Bio-Medico of Rome, Rome, Italy
7National Reference Laboratory of HIV, National Center of Infectious and Parasitic Diseases, Sofia, Bulgaria
8Department of Food Hygiene, Experimental Zooprophylactic Institute of Sardinia, Sassari, Italy
9Emerging Pathogens Institute, University of Florida, Gainesville, FL, USA
1. Introduction
Mayaro virus (MAYV), belongs to Togaviridae family, triggers a febrile arthralgia syndrome close to Dengue and Chikungunya fever. The virus isolated in Trinidad in 1954 is a mosquito-borne alphavirus reported in the South and Central America[1].
The genome is a single-strand RNA of 11.5 kb[2,3]. It has two different open reading frames. The first one translates four non-structuralproteins(NSP1–4),whereasthesecondopenreadingframe encodes a single polyprotein, subsequently being processed to five several proteins: capsid protein (C), two envelope surface glycoproteins (E1 and E2), and two small peptides (E3 and 6k)[4,5].Virus natural cycle involves non-human primates andHaemagogusspp. mosquitoes in tropical areas of South America[6].
MAYV causes Mayaro fever (MF), a neglected endemic syndrome oftropicalAmericas.After7–12dfollowingamosquitobiteasincubation time, patients develop fever, rash, headache, and arthralgia that persist for several weeks[6]. The most prominent symptoms,represented by joint pain and edema, are typical of the acute phase of the disease but can persist chronically for several months[7,8].Even though the disease is not severe and none case of death has been reported, MF cause significant morbidity especially in rural population[9], in rare case also with hemorrhagic manifestations[10].The disease can determine temporary incapacitation to work and hospitalization in some cases[1].
Two different genotypes have been described during MAYV epidemics, the D genotyped prevalently isolated in Brazil and the L genotype with a more wide distribution[1,10]. Reproducing the path of transmission followed by Chikungunya virus (CHIKV), Dengue virus (DENV) and Zika virus (ZIKV) in the Western countries and Indian regions, MAYV has the potentiality to become a global pathogen, due to its transmission mediated by urban mosquitoes such asAedes aegypti[11]. Similarly to CHIKV and ZIKV infection,MAYV epidemics can be undiagnosed during DENV outbreaks and incorrectly misdiagnosed as DENV infection in about 1%of cases[12]. Case definition is clinical definition plus laboratory diagnosis of MAYV[1].
MAYV identification is difficult to be achieved due to the relative short duration of the viremic phase[10]. The available treatment for MF is based on non-steroidal anti-inflammatory medications and chloroquine, and no vaccine or specific antiviral drugs are available at the moment[13]. For this reason, MAYV infection should represent an emerging public health threat, and improved surveillance and preventive measures should be provided to mitigate the already burden on health systems in Latin America so as to limit the spread of the virus infection.
In this study, the genetic diversity of MAYV has been investigated with the aim to estimate the phylogenetic relationships between MAYV strains circulating worldwide. Since MAYV is considered an emergent pathogen, evolutionary analyses have been performed to obtain a comprehensible overview positive-selection analyses and homology modelling has been also performed to evaluate the pathogen evolution and the virus evolution consequences in the protein recognition by host immune response. Homology modelling of MAYV proteins has been applied to allow mapping of sites under pressure and to put forward structural hypotheses about possible consequences of mutations on virus protein recognition by host immune response.
2. Materials and methods
2.1. Sequence dataset
Nine different datasets were assembled, eight for each protein[C, E1, E2, E3, non-structural protein (NSP) 1, NSP2, NSP3, and NSP4] and one for the complete genome, including sequences dated from the year 1955 to the year 2015. All the datasets contained 33 sequences for each protein and the complete genome. All the sequences and complete genome were downloaded by the availability from the NCBI database (http://www.ncbi.nlm.nih.gov/)and all the different genes were analyzed separately. All sequences were aligned using MAFFT software v.7 and manually edited using Bioedit software[14,15]. Modeltest software version 3.7[16] was used to choose the best evolutionary model.
2.2. Phylogenetic signal and maximum likelihood analysis
The phylogenetic signal has been evaluated analyzing randomly 10 000 groups of four sequences (quartets), by the likelihood mapping method using TREE-PUZZLE[17]. The dots inside the equilater triangles correspond to the likelihood of the possible trees.In the triangle, the corners represent the trees with fully resolved topologies, the epicenter area represents the star-like phylogeny,and the areas on the sides correspond to the network-like phylogeny(presence of recombination or conflicting phylogenetic signals).Forsubstitution/saturationanalysisassessment,theXia’stestwasused with the transitions/transversions ratiovs. divergence graph in DAMBE [http://dambe.bio.uottawa.ca/DAMBE/]. The percentage of constant sites and parsimony-info sites were estimated using MEGA7.
The maximum likelihood tree was inferred with the evolutionary model previously selected with ModelTest, by using Phyml[18].The phylogenetic tree was confirmed with the bootstrap analysis(bootstrap values >75%) as reliability test for the branches.
2.3. Selective pressure analysis
Adaptive Evolution Server (http://www.datamonkey.org/) was used to predict the ratio (ω) of non-synonymous to synonymous substitution rates for each codon, with ω>0 indicating diversifying or positive selection[19]. The following selection analyses were conducted according to the best-fit substitution model, also determined within the Server: adaptive branch-site random effects likelihood [aBSREL][20] model for the detection of lineage-specific selection, fast unconstrained Bayesian approximation [FUBAR][21]for inferring site-specific pervasive selection, Bayesian unrestricted test for episodic diversifying selection [BUSTED][21] across the region of interest, and the mixed effects model of evolution [MEME]to identify episodic selection at individual sites[21]. Sites were considered to have been subjected to statistically significant positive or negative selection based on the following cut-offs: likelihoodratio test (LRT)P≤0.05 for BUSTED and aBSREL and posterior probability (PP)] >0.90 for FUBAR, and LRT≤0.05 [withP≤0.01]for MEME. The reference sequence used to trace the amino acid position found under selection was Accession Number: KX496990.1.
2.4. Homology modelling and structural analysis
Homology modeling relied on the software Modeller version 9.17[22]. Template identification was carried out with the programs Blast[23], HHPred[24] or Phyre2[25]. Suitable templates were considered those structures sharing the highest sequence similarity to the target protein, solved at the best resolution available, and with maximum sequence coverage. Sequence alignments were calculated with Clustal Omega[26] and were displayed or edited with the Jalview editor[27]. Ten models of each target protein were built at the highest refinement level and that one showing the lowest value of the Modeller target function, which is indicative of model quality, was considered the representative model. ProsaII[28] and Procheck[29] were utilized for model validation, while protein structure visualization and analysis were carried out with the graphic software PyMOL[30] or Chimera[31]. B-cell epitopes were predicted with a set of publicly available servers[32] listed in Table 1. Proteinprotein interaction site prediction used the meta-PPISP server.

Table 1 B-cell epitope prediction for the Mayaro E1 protein variants.
3. Results
3.1. Phylogenetic analysis and likelihood mapping
As a first step, the phylogenetic signal for all the genomic regions of the MAYV (C, E1, E2, E3, NSP1, NSP2, NSP3, NSP4, and completegenome)bylikelihoodmapping(Figures1A–1I)wastested.The percentage of likelihood points in the central area of the triangles was 12.9% for the first dataset (Capsid gene), 7.5% forE1gene,3.4% forE2gene, 19.9% forE3gene, 3.8% forNSP1gene, 3.1% forNSP2gene, 7.0% forNPS3gene, 9.4% forNSP4gene, and 1.2% for the main dataset, complete genome region. All the datasets showed sufficient phylogenetic signal (30%). All the genetic signal values and phylogenetic information of the MAYV datasets are shown in Table 2. The percentage of the Parsimony-Info sites ranged from 1.74% (E2 dataset)–19.65%(E1dataset);insteadthepercentageoftheconstantsitesrangedfrom73.62%(E1dataset)–81.72%(NSP1dataset).
In the maximum likelihood tree, two main clades (A and B) were highlighted (Figure 2). The clade A included six sequences, sampled from 1955 to 2015, of which five from Brazil and one from Haiti.The Brazilian sequence sampled in 2014 clusters with the Haitian sequence sampled in 2015 with high bootstrap value (100%),suggesting a very close phylogenetic relationship. The clade B included the remaining 27 sequences sampled in the Central and Southern America (Bolivia, Brazil, French Guiana, Peru, Trinidad and Tobago and Venezuela) from 1957 to 2013. Inside the clade B, there is a statistically supported clade (B1) where one Peruvian sequence sampled in 2010 can be considered outgroup of the clade B1. Inside the clade B1, there are several statistically supported clusters, grouping mostly according the geographic location.
3.2. Selective pressure analysis
Genetic variability was determined by nucleotide sequencing of fragments ranging from 198 nt for the E3 protein to 2 394 nt for the NSP2 protein. The α parameter of the γ distribution for all the proteins analyzed was <1, demonstrating L-shape characteristic of the distribution and proposing that rate heterogeneity of nucleotide substitution is across sites. Selective pressure analyses performed considering all the analyzed genomic regions (C,E1,E2,E3,NSP1,NSP2, andNSP4) highlighted eight sites (107, 215, 243, 11, 231,497, 251, and 64) under strong negative selection (by using FUBAR)indicated by ω<0, suggesting high degree conservation in the genomic region analyzed (Table 2). Using BUSTED, it has been highlighted episodic diversifying selection only in E3 and NSP4.No branch-specific episodic diversifying selection was found using aBSREL. Few sites with episodic diversifying selection were found in E1, NSP1 and NSP3 with LRTP≤ 0.01. There was one on each gene: amino acidic position 300 in E1, amino acidic position 242 in NSP1 and amino acidic position 31 in NSP3 (using MEME).
3.3. Homology modeling and structure analysis
Homology model of the MAYV E1 protein (GenBank code ANY58848) was built using the templates represented by the E1 envelope proteins from Semliki Forest virus (SFV) (PDB code 2ALA, resolution 3.0 Å) and CHIKV (PDB code 3N44, 2.35 Å). The sequence alignment on which the modeling was based is displayed in Figure 3. MAYV E1 shares 76% and 60% sequence identity with SFV and CHIKV proteins, respectively. Figure 4 reports the superposition between the best MAYV E1 model and the structures of the template proteins, and it also shows the location of sequence position 300 which was found to be under selective pressure.This site is located in the domain III of the E1 protein within a β-strand of the immunoglobulin-like fold, before a conserved Cys involved in a disulfide bond. A multiple sequence alignment of a set of 58 homologous E1 proteins from various alphaviruses shows that position 300 can hold Ser (with frequency 58%), Glu(18%), Lys (12%), Thr (5%), Leu (3%) or Ala (1%). Interestingly,MAYV E1 displays a two-residue deletion in correspondence of positions344–345oftheSFVandCHIKVE1proteins(Figure3). This deletion occurs within a β-strand of the β-barrel of the immunoglobulin fold and may induce a partial local conformational rearrangement of the MAYV E1 loop encompassed by the sequence positions346–349ofdomainIII.Epitopeprediction(Table2)ofprotein E1 variants which possess Thr or Leu at position 300 was carried out using 11 methods listed in Table 1[32]. Epitope prediction is quite inaccurate and for that reason a consensus approach is generally recommended[32]. Overall, although no clear consensus is evident, position 300 is predicted to be part of a region possessing potential epitope propensity. In fact, the 5thand 6thmethods out of the 11 prediction methods employed indicate the occurrence of a potential epitope with Leu or Thr at position 300, respectively (Table 2). Since the region bearing Thr is predicted as a potential epitope by six methods, it may be conceived that the occurrence of this residue can attribute a marginally higher epitope propensity. No suitable template was found for the protein NSP1 and for that reason no homology model could be built in our study. Modelling of the macrodomain of MAYV NSP3 protein (Genbank code ALJ56198.1) utilized the following templates: a fragment of the NSP from Sindbis virus (PDB code 4GUA, resolution 2.85 Å); macrodomain from CHIKV (3GPG,1.65 Å) and macrodomain of venezuelan equine encephalitis virus in complex with ADP-ribose (3GQO, 2.60 Å). The complex between MAYV NSP3 and ADP-ribose was predicted following the geometry of the complex reported in 3GQO. MAYV NSP3 shares 52%, 66% and 56% sequence identity with 4GUA, 3GPG, and 3GQO, respectively.According to the homology model, sequence position 31 of the MAYV NSP3 is predicted at the mouth of the macrodomain binding site of the ADP-ribose (Figure 5) in proximity of residues (for example Asn24 and Val33) considered to be important for ADP-ribosylhydrolase activity in the homologous CHIKV NSP3 macrodomain[44]. Prediction of protein-protein interaction sites attributed position 31 a high interaction propensity (Figure 5).

Figure 1. Likelihood mapping of all datasets.

Table 2 Evidence of selection pressure among codon sites in all the coding regions analyzed capsid, E1, E2, E3, NSP1, NSP2, NSP3 and NSP4 genes.

Figure 2. Maximum likelihood tree of Mayaro virus.

Figure 4. Structural superposition among the MAYV E1 homology model(orange cartoon) and the homologous proteins from Semliki Forest virus(grey) and Chikungunya virus (green).
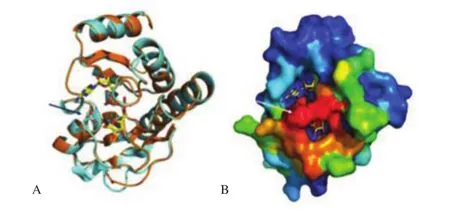
Figure 5. Prediction of structural protein position.

Figure 3. Sequence alignment among the Mayaro E1 protein (labeled by HAITI) and the sequences of the E1 envelope proteins from Semliki Forest virus(indicated by 2ALA) and Chykungunya virus (3N44 chain F).
4. Discussion
MAYV has been reported in several tropical countries in the Central and South America, as Brazil[1,45]. MAYV has the potentiality to become a global pathogen, being its transmission mediated by urban mosquitoes such asAedes aegypti, as happened in the recent past for CHIKV[46,47]. This analogy can also be supported by similar clinical symptoms. Indeed the illness, which can cause chills, malaise,headache, stomach pains and joint pains, is transmitted by that same vectorAedes aegyptimosquito which transmits ZIKV, DENV, yellow fever virus and CHIKV. As recently reported in CHIKV and ZIKV epidemics, MAYV outbreaks can pass undiagnostigsate during DENV epidemics[46,48]. Indeed the clinical symptoms of Mayaro are so similar to Chikungunya and several other ailments that it is easy to be misdiagnosed. Moreover dual infections from these viruses can further complicate accurate diagnosis. MAYV is the one of the long list of recently emerging viral pathogens that has received global attention due to various outbreaks in tropical areas and its rapid spread in several countries. Maximum likelihood analysis confirmed the MAYV previous presence in Haiti and successful spread to areas such as the Caribbean and USA[49].
Selective pressure analysis performed in this study revealed a strong presence of negatively selected sites in all genomic regions analyzed, suggesting a probable exclusion of deadly polymorphisms in functionally genes. At the same time, this analysis highlighted a few sites with episodic diversifying selection in E1, NSP1 and NSP3 with LRTP≤0.01 located in the amino acidic position 300 in E1,amino acidic position 242 in NSP1 and amino acidic position 31 in NSP3.
The contemporary lack of presence of high percentage of positively selected sites also suggests and enforces the idea of highly adapted phenotypes. Probably, the alternation cycle between arthropod vector and human and no human hosts may impose a sort of barrier to nonsynonymous mutations in important genes. Amino acid conservancy within MAYV sequences can be considered in combination with potential immune escape amino acid mutations. The homology modeling analysis showed localized high mutation frequency in the structural and non-structural proteins (NS3 and NS5) corresponding to a considerable alteration in the protein stability.
The position of the site 300 under positive selective pressure has been mapped onto the predicted three-dimensional structure of the MAYV E1 protein. In particular, model inspection indicates that this site is located in the domain III of the E1 protein within a β-strand of the immunoglobulin-like fold, before a conserved Cys involved in a disulfide bond[50,51]. In the E1 envelope protein of Sindbis virus, belonging to theAlphavirusgenus, the position equivalent to the MAYV E1 300 was proven to be part of a set of transitional epitopes that become accessible only after virus particle exposures to agents, such as heat, low pH and the like[50,51]. Change in epitope accessibility was observed also upon virus particle exposure to susceptible cells, which suggests that a conformational modification of the envelope proteins takes place during cell binding.Moreover, the structure site of the SFV E1 protein equivalent totheMAYVLeu300isinvolvedinE1–E1contactwithinthevirus protein shell[52]. The high similarity between the MAYV E1 and the homologous protein sequences from other alphaviruses suggests that the position 300 may have properties similar to those attributed in the other viruses. Moreover, within the limits of intrinsic inaccuracy, B-cell epitope prediction suggests that the region surrounding the position 300 might have an immunogenic potential. Interestingly, Thr at position 300 apparently may attribute a higher epitope potential than the presence of Leu at the same position. A multiple sequence alignment of a set of 58 homologous E1 proteins from various alphaviruses shows that position 300 can in other viruses hold other residues mostly polar such as Ser (with frequency 58%). Furthermore, MAYV E1 displays a two-residue deletion in correspondence of positions 344–345 of the SFV and CHIKV E1 proteins. This deletion occurs within a β-strand of the β-barrel of the immunoglobulin fold and may induce apartial local conformational rearrangement of the MAYV E1 loopencompassed by the sequence positions 346–349 of domain III.All these observations hint at the potential existence of an ongoing fine process of MAYV E1 protein adaptation to cope with host immunological surveillance.
MAYV protein NSP3 contains a N-terminal domain homologous to the highly conserved macrodomain[53], which, in CHIKV, has been demonstrated to possess ADP-ribosylhydrolase activity essential for virus replication and virulence[44]. According to the homology model, the position 31 found to be under selective pressure is located in the edge of the ADP-ribose binding site and is predicted to possess a high potential of protein-protein interaction. It may be conceived that this position is involved in the recognition of the intracellular targets of the MAYV macrodomain and consequently is subjected to functional constraints. Currently, no licensed vaccines are available for MF, and the only control strategy is based on human exposure to potential vectors reduction. Two attempts of vaccine are described in literature, one in human diploid cells and the other in murine models[54]. A protective vaccine could decrease the potential risk of MAYV urbanization through adaptative mutation that enhance the vectorial ability of transmission, as happened with CHIKV[55] and could be of great impact on public health.
MAYV has many undiscovered features of concern, and since it was first isolated in Trinidad in 1954, it has been linked with limited epidemics in Southern America[56] near the Amazon rainforest.MAYV infection syndrome includes a group of symptoms such as arthralgias, fever, headache, myalgias, rash, and sporadically nausea and vomiting[57]. MAYV infections are underdiagnosed because of a possible misleading with other mosquito-borne virus infections,especially dengue fever, endemic in common areas. Recently,CHIKV has added disorientation in the diagnosis, especially due to the prolonged arthralgia connected with CHIKV and MAYV cases[56]. Our findings confirmed that MAYV is spreading in Brazil since its isolation in the 1950s[49]. A serious concern arises from the easy interaction and proclivity for MAYV/DENV co-infections. That phenomenon should be the subject of careful studies and include other arboviruses from the same geographic regions to clarify the nature of the MAYV epidemic or other similar pathogens. In addition, in light of these considerations, conducting molecular and evolutionary studies are important to reveal the potential abilities of this virus to trigger a significant epidemic, and help to elucidate the MAYV way of transmission byAedesandHaemagogusspp.mosquitoes.
Conflict of interest statement
We declare that we have no conflict of interest.
[1] Mota MTdO, Ribeiro MR, Vedovello D, Nogueira ML. Mayaro virus: A neglected arbovirus of the Americas.Fut Virol2015; 10: 1109-1122.
[2] Gouandijka-Vasilache I, Manirakiza A, Gody JC, Banga-Mingo V,Kongombe OO, Esona MD, et al. Rotavirus epidemiology in Bangui,Central African Republic, 2008.Emerg Infect Dis2014; 20(7): 1254-1255.
[3] Lavergne A, de Thoisy B, Lacoste V. Mayaro virus complete nucleotide sequence and phylogenetic relationships with other alphaviruses.Virus Res2006; 117(2): 283-290.
[4] Powers A, Huang H, Roehring J, Strauss E, Weaver S. FamilyTogaviridae. In: King AMQ, Lefkowitz E, Adams MJ, Carstens EB.(editors)Virus taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses. San Diego: Elsevier Science; 2011, p. 1103-1110.
[5] Hallengard D, Kakoulidou M, Lulla A. Novel attenuated Chikungunya vaccine candidates elicit protective immunityin C57BL/6 mice.J Virol2014; 88(5): 2858-2866.
[6] Azevedo RS, Silva EV, Carvalho VL, Rodrigues SG, Nunes-Neto JP,Monteiro H. Mayaro fever virus, Brazilian Amazon.Emerg Infect Dis2009; 15(11): 1830-1832.
[7] Vasconcelos PF, Calisher CH. Emergence of human arboviral diseases in theAmericas,2000–2016.Vector Borne Zoonot Dis2016; 16(5): 295-301.
[8] Neumayr A, Gabriel M, Fritz J. Mayaro virus infection in traveller returning from Amazon Basin, northern Peru.Emerg Infect Dis2012;18(4): 695-696.
[9] Pinheiro FP, Freitas RB, Travassos da Rosa JF, Gabbay YB, Mello WA,LeDuc JW. An outbreak of Mayaro virus diseasein Belterra, Brazil. I.Clinical and virological findings.Am J Trop Med Hyg 1981; 30(3): 674-681.
[10] Mourao MP, Bastos Mde S, de Figueiredo RP. Mayaro fever in the city of ManausBrazil,2007–2008.Vector Borne Zoonot Dis2012; 12(1): 42-46
[11] Long KC, Ziegler SA, Thangamani S. Experimental transmission of Mayaro virus byAedes aegypti.Am J Trop Med Hyg2011; 85(4): 750-757.
[12] Forshey BM, Guevara C, Laguna-Torres VA, Cespedes M, Vargas J,Gianella A. Arboviral etiologies of acute febrile illnesses in Western SouthAmerica,2000–2007.PLoS Negl Trop Dis2010; 4(8): e787.
[13] Taubitz W, Cramer JP, Kapaun A. Chikungunya fever intravelers: Clinical presentation and course.Clin Infect Dis2007; 45(1): e1-4.
[14] Katoh K, Kuma K, Toh H, Miyata T. MAFFT version 5: Improvement in accuracy of multiple sequence alignment.Nucleic Acids Res2005; 33(2):511-518.
[15] Hall TA. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT.Nucl Acids Symp1999; 41:95-98.
[16] Posada D, Crandall KA. MODELTEST: Testing the model of DNA substitution.Bioinformatics1998; 14(9): 817-818.
[17] Strimmer K, von Haeseler A. Likelihood-mapping: A simple method to visualize phylogenetic content of a sequence alignment.Proc Natl Acad Sci USA1997; 94(13): 6815-6819.
[18] Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0.Syst Biol2010;59(3): 307-321.
[19] Pond SL, Frost SD, Muse SV. HyPhy:hypothesis testing using phylogenies.Bioinformatics2005; 21(5): 676-679.
[20] Kosakovsky Pond SL, Frost SD. Not so different after all: A comparison of methods for detecting amino acid sites under selection.Mol Biol Evol2005; 22(5): 1208-1222.
[21] Murrell B, Moola S, Mabona A, Weighill T, Sheward D, Kosakovsky Pond SL, et al. FUBAR: A fast, unconstrained Bayesian approximation for inferring selection.Mol Biol Evol2013; 30(5): 1196-1205.
[22] Webb B, Sali A. Comparative protein structure modeling using MODELLER.Curr Protoc Bioinformatics2016; 54: 5.6.1-5.6.37.
[23] Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W,et al. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs.Nucleic Acids Res1997; 25(17): 3389-3402.
[24] Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction.Nucleic Acids Res2005; 33:W244-248.
[25] Kelley LA, Sternberg MJ. Protein structure prediction on the Web: A case study using the Phyre server.Nature Protoc2009; 4(3): 363-371.
[26] Sievers F, Higgins DG. Clustal omega.Curr Protoc Bioinformatics2014;48: 3.
[27] Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview Version 2--a multiple sequence alignment editor and analysis workbench.Bioinformatics2009; 25(9): 1189-1191.
[28] Wiederstein M, Sippl MJ. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins.Nucleic Acids Res2007; 35: W407-W410.
[29] Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR.J Biomol NMR1996; 8(4): 477-486.
[30] Schroedinger LLC. The PyMOL Molecular Graphics System 2015;Version 1.8; 2015.
[31] Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM,Meng EC, et al. UCSF Chimera--a visualization system for exploratory research and analysis.J Comput Chem2004; 25(13): 1605-1612.
[32] Potocnakova L, Bhide M, Pulzova LB. An introduction to B-cell epitope mapping andin silicoepitope prediction.J Immunol Res2016; 2016:6760830.
[33] Saha S, Raghava GP. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network.Proteins2006; 65(1): 40-48.
[34] Chen J, Liu H, Yang J, Chou KC. Prediction of linear B-cell epitopes using amino acid pair antigenicity scale.Amino Acids2007; 33(3): 423-428.
[35] Larsen JE, Lund O, Nielsen M. Improved method for predicting linear B-cell epitopes.Immunome Res2006; 2: 2
[36] Yao B, Zhang L, Liang S, Zhang C. SVMTriP: A method to predict antigenic epitopes using support vector machine to integrate tri-peptide similarity and propensity.PLoS One2012; 7(9): e45152.
[37] Rubinstein ND, Mayrose I, Martz E, Pupko T. Epitopia: A web-server for predicting B-cell epitopes.BMC Bioinformatics2009; 10: 287.
[38] Sweredoski MJ, Baldi P. PEPITO: Improved discontinuous B-cell epitope prediction using multiple distance thresholds and half sphere exposure.Bioinformatics2008; 24(12): 1459-1460.
[39] Kringelum JV, Lundegaard C, Lund O, Nielsen M. Reliable B cell epitope predictions: Impacts of method development and improved benchmarking.PLoS Comput Biol2012; 8(12): e1002829.
[40] Liang S, Zheng D, Zhang C, Zacharias M. Prediction of antigenic epitopes on protein surfaces by consensus scoring.BMC Bioinformatics2009; 10: 302.
[41] Liang S, Zheng D, Standley DM, Yao B, Zacharias M, Zhang C. EPSVR and EPMeta: Prediction of antigenic epitopes using support vector regression and multiple server results.BMC Bioinformatics2010; 11: 381.
[42] Ponomarenko J, Bui HH, Li W, Fusseder N, Bourne PE, Sette A, et al. ElliPro: A new structure-based tool for the prediction of antibody epitopes.BMC Bioinformatics2008; 9: 514.
[43] Ansari HR, Raghava GP. Identification of conformational B-cell epitopes in an antigen from its primary sequence.Immunome Res2010; 6: 6
[44] McPherson RL, Abraham R, Sreekumar E, Ong SE, Cheng SJ, Baxter VK, et al. ADP-ribosylhydrolase activity of Chikungunya virus macrodomain is critical for virus replication and virulence.Proc Natl Acad Sci U S A2017; 114(7): 1666-1671.
[45] Vieira CJ, Silva DJ, Barreto ES. Detection of Mayaro virus infections during a dengue outbreak in Mato Grosso,Brazil.Acta Trop2015; 147:12-16.
[46] Lo Presti A, Cella E, Angeletti S, Ciccozzi M. Molecular epidemiology,evolution and phylogeny of Chikungunya virus: An updating review.Infect Genet Evol2016; 41: 270-278.
[47] Furuya-Kanamori L, Liang S, Milinovich G. Co-distribution and coinfection of chikungunya and dengue viruses.BMC Infect Dis2016; 16:84.
[48] Giovanetti M, Milano T, Alcantara LC, Carcangiu L, Cella E, Lai A,et al. Zika virus spreading in South America: Evolutionary analysis of emerging neutralizing resistant Phe279Ser strains.Asian Pac J Trop Med2016; 9(5): 445-452.
[49] Mavian C, Rife BD, Dollar JJ, Cella E, Ciccozzi M, Prosperi MCF, et al.Emergence of recombinant Mayaro virus strains from the Amazon basin.Sci Rep2017 7(1): 8718.
[50] Voss JE, Vaney MC, Duquerroy S, Vonrhein C, Girard-Blanc C, Crublet E, et al. Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography.Nature2010; 468(7324): 709-712.
[51] Mukhopadhyay S, Zhang W, Gabler S, Chipman PR, Strauss EG,Strauss JH, et al. Mapping the structure and function of the E1 and E2 glycoproteins in alphaviruses.Structure2006; 14(1): 63-73.
[52] Meyer WJ, Johnston RE. Structural rearrangement of infecting Sindbis virions at the cell surface: Mapping of newly accessible epitopes.J Virol1993; 67(9): 5117-5125.
[53] Roussel A, Lescar J, Vaney MC, Wengler G, Wengler G, Rey FA.Structure and interactions at the viral surface of the envelope protein E1 of Semliki Forest virus.Structure2006; 14(1): 75-86.
[54] Rack JG, Perina D, Ahel I. Macrodomains: Structure, function, evolution,and catalytic activities.Annu Rev Biochem2016; 85: 431-454.
[55] Robinson DM, Cole FE Jr, McManus AT, Pedersen CE Jr. Inactivated Mayaro vaccine produced in human diploid cell cultures.Mil Med1976;141(3): 163-166.
[56] Tsetsarkin KA, Chen R, Leal G, Forrester N, Higgs S, Huang J, et al.Chikungunya virus emergence is constrained in Asia by lineage-specific adaptive landscapes.Proc Natl Acad Sci U S A2011; 108(19): 7872-7877.
[57] Tesh RB, Watts DM, Russell KL, Damodaran C, Calampa C, Cabezas C, et al. Mayaro virus disease: An emerging mosquito-borne zoonosis in tropical South America.Clin Infect Dis1999; 28(1): 67-73.
杂志排行

Asian Pacific Journal of Tropical Medicine的其它文章
- Therapeutic role of Ricinus communis L. and its bioactive compounds in disease prevention and treatment
- Dietary isoflavones, the modulator of breast carcinogenesis: Current landscape and future perspectives
- Nacre extract prevents scopolamine-induced memory deficits in rodents
- Evaluating therapeutic potential of coriander seeds and leaves(Coriandrum sativum L.) to mitigate carbon tetrachloride-induced hepatotoxicity in rabbits
- Andrographolide inhibits chikungunya virus infection by up-regulating host innate immune pathways
- Effects of some common additives on the antimicrobial activities of alcohol-based hand sanitizers