SAS诱导氧化应激时人卵巢癌顺铂敏感细胞与耐药细胞死亡方式的差异
2018-04-24高玮男张雪双孙连坤于春艳刘亚男
高玮男,张雪双,孙连坤,于春艳*,刘亚男*
(1.吉林大学临床医学院,吉林 长春130021;2.北华大学基础医学院病理学教研室;3.吉林大学基础医学院病理生理学系)
Bcl2抑制剂ABT737能够诱导SKOV3/DDP细胞发生凋亡,但不能完全抑制SKOV3/DDP 细胞增殖[1],提示耐药细胞不仅存在凋亡逃逸,还有可能存在其他形式的细胞死亡。氧化应激时产生的ROS(reactive oxygen species)可能是细胞凋亡(apoptosis)、铁死亡(ferroptosis)以及程序性坏死(necroptosis)的重要传播者和执行者[2-4]。然而根据细胞的状态和诱导的条件不同,ROS介导何种细胞死亡方式目前尚不清楚。本研究利用SAS(柳氮磺吡啶),谷氨酸/胱氨酸转运体(glutamate/cystine antiporter,xc-)抑制剂,导致细胞内氧化应激,旨在观察在氧化应激条件下,卵巢癌亲本细胞SKOV3和耐药细胞SKOV3/DDP能否发生程序性坏死、铁死亡等非凋亡性细胞死亡途径,同时对比两种细胞死亡途径的差异,为寻找卵巢癌耐药的分子机制提供实验依据和研究靶点。
1 材料与方法
1.1药品、主要试剂和仪器IMDM 培养基、胰蛋白酶产自美国 Hy Clone 公司;1640培养基、DMEM培养基、新生牛血清产自美国 Gibco 公司;细胞培养板产自 Corning 公司;细胞培养皿产自Thermo公司;0.22 μm 滤膜、产自美国 Invitrogen 公司;Sulfasalazine (SAS)、MTT、坏死抑制剂Nec-1、铁死亡抑制剂Fer-1产自美国 Sigma公司。RTCA DP(Real time cell analysis)来自美国罗氏公司。MLKL及磷酸化MLKL抗体购自Abcam公司。
1.2细胞培养及分组卵巢癌SKOV3、SKOV3/DDP细胞系均来自吉林大学病理学与病理生理学系研究室。卵巢癌SKOV3细胞用含有10%胎牛血清的1640培养液培养;卵巢癌SKOV3/DDP细胞在培养皿中,含有10%胎牛血清的IMDM培养液培养,卵巢癌SKOV3/DDP细胞每次传代前加入1 μg/ml顺铂,以维持卵巢癌SKOV3/DDP细胞的耐药性。细胞置于CO2培养箱(37℃,5% CO2)培养,隔2-3d传代1次,接种后24 h于对数生长期的细胞用于实验。实验分组为对照组(Con),SAS各剂量组。 应用铁死亡抑制剂Fer-1,实验分组为对照组,SAS组,Fer-1组,Fer-1与SAS联合作用组。应用程序性坏死抑制剂Nec-1,实验分组为对照组,SAS组,Nec-1组,Nec-1与SAS联合作用组。
1.3MTT法测定细胞增殖抑制率SKOV3和SKOV3/DDP细胞接种于96孔培养板,每组细胞5复孔,于培养结束前4 h加入10 μl的MTT,培养结束后吸出去培养基,每孔加入DMSO 150 μl,振荡混匀15 min,使结晶完全溶解,用酶联免疫检测仪于490 nm波长处测定其吸光度(A)值。对照组细胞增殖抑制率为0%,其余各组细胞增殖抑制率=(对照组A值-实验组A值/对照组A值)×100%。
1.4RTCA实时无标记细胞功能分析法实时连续分析细胞
先用培养液调零,然后按照每孔100 μl的培养液中含有8×103~1.5×104个细胞的浓度接种至金孔板,将金孔板嵌入RTCA的连接仪器,然后在37℃,含有5% CO2的细胞培养箱内继续培养。当细胞长至对数生长期时,将原液弃掉,加入配置好的药物,再将金孔板嵌入RTCA的连接仪器,然后在37℃,含有5% CO2的细胞培养箱内继续培养。观察细胞的实时生长曲线,当细胞的生长曲线达到要求时,结束培养,并利用RTCA Data Analysis Software 1.0软件分析结果。
1.5Westernblotting法检测细胞中MLKL和磷酸化MLKL蛋白表达用RIPA裂解液提取细胞总蛋白,以β-actin的水平作为等量蛋白质上样对照,取50 μg蛋白质样品进行SDS-PAGE电泳,转至硝酸纤维素膜上;5%奶粉室温封闭2 h后,用含0.01% Tween20的TBS缓冲液漂洗3次,每次10 min;加入相应的抗体(1∶1000),4℃孵育过夜,TBST漂洗3次后加入相应的辣根过氧化物酶标记的二抗(1∶1000),37℃摇床温育2 h;TBST漂洗3次,每次10 min;DAB显色,凝胶图像分析系统拍照,同时以β-actin为内参照,进行蛋白表达分析,蛋白表达水平以吸光度(A)值的比值表示,按照下列公式计算蛋白表达水平,并进行统计分析。蛋白表达水平=每个样本条带的A值/β-actin的A值。

2 结果
2.1SAS对SKOV3和SKOV3/DDP细胞生存率的影响
MTT实验结果显示,与对照组(Con)比较,不同浓度SAS作用 SKOV3细胞和SKOV3/DDP细胞,细胞生存率均降低(P<0.05);与SKOV3细胞比较,相同浓度的SAS作用SKOV3/DDP细胞,细胞生存率降低程度不明显,表明SKOV3 和SKOV3/DDP细胞对SAS的敏感性不同,SKOV3/DDP细胞对SAS耐受。其中,0.5 mM SAS作用SKOV3细胞,细胞生存率为(80±3)%,SAS 2 mM作用SKOV3/DDP细胞,细胞生存率为(80±4)%,这两个剂量作为后续实验所用剂量。见图1。
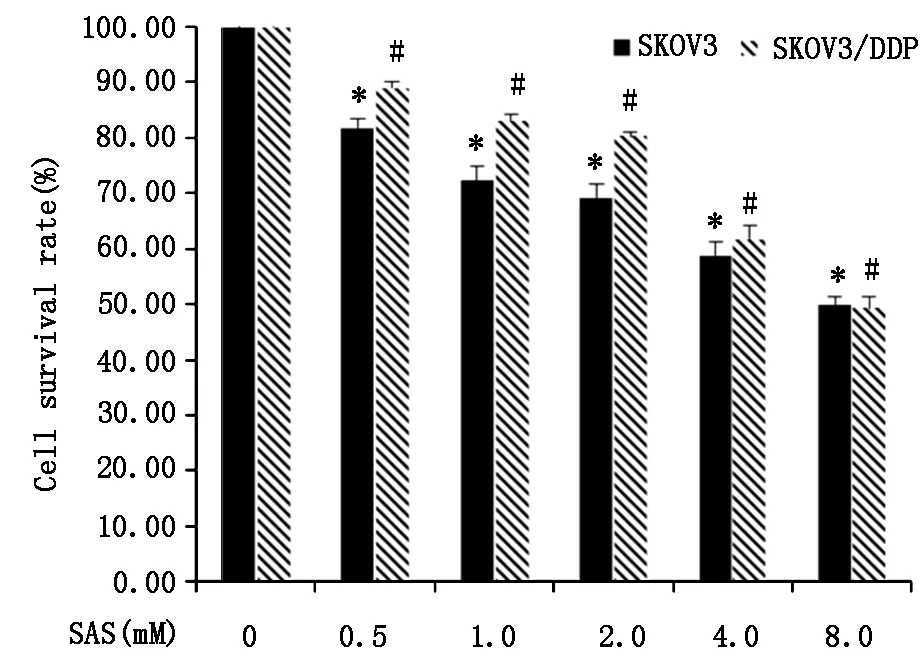
图1不同浓度SAS对SKOV3、SKOV3/DDP细胞生存率的影响n=3,*和#:comparedwithSASgroup
2.2MTT法检测铁死亡抑制剂Fer-1对SAS抑制SKOV3和SKOV3/DDP细胞生存率的影响
SKOV3 MTT结果显示,与对照组(Con)比较,SAS作用SKOV3细胞组生存率为(79.5±1.1)%;与SAS 0.5 mM组比较,Fer-1各个浓度与SAS联合作用24 h组SKOV3生存率没有增加(P>0.05);与对照组比较,不同浓度的Fer-1组生存率没有差异(P>0.05)。见图2。
SKOV3/DDP MTT结果显示,与对照组(Con)比较,SAS生存为(78.9±1.6)%。与SAS 2mM组比较,Fer-1 1.0 μM和2.0 μM与SAS 2 mM联合作用,SKOV3/DDP细胞生存率增加(P<0.05)。与Con组比较,单独Fer-1各浓度组细胞生存率没有统计学意义(P>0.05)。见图2。
2.3RTCA法检测铁死亡抑制剂Fer-1对SAS抑制SKOV3和SKOV3/DDP细胞生存率的影响

图2MTT法检测不同浓度Fer-1与SAS对SKOV3和SKOV3/DDP细胞生存率的影响(n=3)
SKOV3细胞RTCA法结果显示,与对照组(Con)比较,SAS 0.5 mM作用SKOV3细胞生存率曲线降低;与SAS组比较,Fer-1各个浓度与SAS联合24 h SKOV3生存率曲线降低。见图3。
SKOV3/DDP细胞RTCA法结果显示,与对照组(Con)比较,SAS 2 mM组生存率曲线降低,与SAS 2 mM组比较,Fer-1 1.0 μM和2.0 μM与SAS 2 mM联合作用仅在24 h前,SKOV3/DDP细胞生存率曲线升高(P<0.05)。表明Fer-1作用24 h前能挽救SAS对SKOV3/DDP细胞生存率的抑制作用。见图3。

图3RTCA法检测Fer-1与SAS联合作用对SKOV3和SKOV3/DDP细胞生存率的影响(n=3)
2.4RTCA法检测程序性坏死抑制剂Nec-1对SAS抑制SKOV3和SKOV3/DDP生存率的影响
SKOV3细胞RTCA结果显示,与对照组(Con)比较,SAS 0.5 mM生存率曲线降低,与SAS 0.5 mM组比较,Nec-1 25 μM与SAS联合作用组,随着作用时间延长SKOV3细胞生存率曲线均低于SAS组的生存率曲线(P>0.05)。SKOV3/DDP细胞RTCA结果显示,Nec-1 5 μM与SAS 2 mM联合作用持续36 h,SKOV3/DDP细胞生存率曲线高于SAS组的生存率曲线,有统计学意义(P<0.05)。见图4。

图4RTCA法检测Nec-125μM与SAS联合作用对SKOV3和SKOV3/DDP细胞生存率的影响(n=3)
2.5Westernblotting法检测程序性坏死标志蛋白的表达水平
Western bloting结果显示在SAS作用卵巢癌SKOV3/DDP细胞12 h、24 h时,程序性坏死相关标志蛋白磷酸化MLKL蛋白(P-MLKL)表达增加(P<0.05);而在SAS作用卵巢癌SKOV3细胞的各个时间点,P-MLKL表达没有变化(P>0.05)。
3 讨论
耐受顺铂是卵巢癌治疗的瓶颈。尽管凋亡的诱导是最为广泛使用的抗肿瘤策略,但由于化疗药物往往存在广泛的凋亡缺陷、多药耐药等难以克服的问题,研究程序性坏死(necroptosis)、铁死亡(ferroptosis)等非凋亡性程序性细胞死亡途径更有意义[5-7]。本研究通过铁死亡抑制剂以及程序性坏死抑制剂,利用实时无标记细胞功能分析法等方法证明SAS诱导卵巢癌亲本细胞SKOV3和耐药细胞SKOV3/DDP发生程序性坏死和铁死亡途径具有差异,与非耐药细胞比较,耐药细胞存在非凋亡性细胞死亡途径。

柳氮磺吡啶(Sulfalsalazine,SAS)是Xc-系统抑制剂,抑制胱氨酸/谷氨酸反向转运体不仅会影响胱氨酸依赖的谷胱甘肽合成,还会抑制胱氨酸的跨膜穿梭,这两种作用都会损坏细胞的抗氧化系统,增加ROS 的积累,在机体的抗氧化应激中发挥关键作用[8]。2012年,Dixon 等[9,10]新提出了一种叫做铁死亡(ferroptosis)的铁依赖性的细胞死亡形式。铁死亡的作用机制可能与谷氨酸的氧化毒性类似,SAS等药物通过胱氨酸/谷氨酸转运体抑制胱氨酸的吸收,从而破坏了细胞内氧化还原平衡的调节并且最终形成依赖于铁离子的氧化性死亡。也已证实SAS诱导顺铂耐受的肺小细胞癌细胞发生凋亡[11],SAS诱导神经胶质瘤细胞U251发生凋亡及自噬[12]。
细胞程序性坏死(Necroptosis),又称为坏死性凋亡,是一种既受死亡信号调控、又出现坏死样结构特点的死亡形式,是一种可被坏死抑制剂Necrostain-1( Nec-1) 抑制的新细胞死亡方式,并且由机体内一系列信号转导因子如RIP1、RIP3、MLKL 等高度调控的过程[13]。研究发现[14],ROS 的大量产生和积聚是RIP3 蛋白依赖的,而且糖原磷酸化酶(PYGL)、谷氨酰胺合成酶(GLUL)和谷氨酸脱氢酶(GLUD1)参与调节。过量的ROS 使得线粒体膜的通透性改变诱发细胞程序性坏死。
本研究利用SAS诱导氧化应激,再分别联合铁死亡抑制剂Fer-1和程序性坏死抑制剂Nec-1,结果显示Fer-1及Nec-1与SAS联合作用SKOV3细胞,均不能挽救SAS对SKOV3细胞生存率的抑制作用。与SKOV3细胞比较,Fer-1作用24 h前挽救SAS对SKOV3/DDP细胞生存率的抑制作用,24 h后不能挽救SAS对SKOV3/DDP细胞生存率的抑制作用;Nec-1能全程性挽救SAS对SKOV3/DDP细胞生存率的抑制作用,表明耐药细胞(SKOV3/DDP)均存在非凋亡性细胞死亡方式,即铁死亡以及程序性坏死途径,而非耐药细胞(SKOV3)则不存在非凋亡性细胞死亡方式,推测可能只存在细胞凋亡途径。接下来我们从蛋白水平上进一步证明。由于RIP1-RIP3 程序性坏死小体形成的必备条件之一,其作用底物为混合谱系激酶结构域样蛋白(MKLK)。MKLK 是程序性坏死通路中RIP3 下游路径中的一个关键酶。因此我们利用Western blotting法检测磷酸化MLKL蛋白表达情况。结果显示,与SAS作用0h比较,SAS作用不同时间点,SKOV3细胞磷酸化MLKL蛋白表达没有变化,而SKOV3/DDP细胞中磷酸化MLKL蛋白表达增强。本研究对比观察到非耐药细胞及耐药细胞在氧化应激条件下细胞死亡方式的差异,而且实时动态观察到,氧化应激在不同时程时,铁死亡与程序性坏死途径所占据的比重不同,在氧化应激早期可见铁死亡及程序性坏死,随着氧化应激时间延长,只存在程序性坏死。
Zhang 等人[15]发现,不表达RIP3 基因的A细胞(一种NIH-3T3 细胞系)在肿瘤坏死因子TNF-α诱导下发生凋亡,而表达RIP3 基因的N 细胞(另一种NIH-3T3 细胞系)却在TNF-α诱导下发生程序性坏死,表明RIP3在细胞程序性坏死与凋亡中的重要开关作用。由此可见,细胞死亡机制间构成了一个复杂的网络系统,凋亡、自噬、程序性坏死等三种程序性坏死以及坏死,在非耐药细胞及耐药细胞中所占比例可能与肿瘤细胞本身状态、肿瘤生存环境、化疗药物作用时间长短及损伤程度等因素密切相关。这四种死亡机制均参与化疗药物诱导肿瘤细胞死亡过程,且四者之间可能相互转换、相互制约,在非耐药细胞中可能某种机制占主导,但在耐药细胞中可能共同参与化疗药物作用。
结合课题组前期研究结果,多功能蛋白P62蛋白在非耐药细胞(SKOV3)中低表达,而在耐药细胞(SKOV3/DDP)中高表达,根据两种细胞死亡方式的不同,提示P62蛋白可能与肿瘤细胞的程序性坏死途径有关。今后的研究中需要重点关注凋亡、程序性坏死和自噬在肿瘤化疗药物耐受中的作用,调控细胞死亡路径之间的转换将很好地拓宽治疗恶性肿瘤的范围。
参考文献:
[1]Xie Q,Su J,Jiao B,et al.ABT737 reverses cisplatin resistance by regulating ER-mitochondria Ca2+signal transduction in human ovarian cancer cells[J].Int J Oncol,2016,49(6):2507.
[2]关 鹏,石振华,李亚青,等.铁死亡:一种新的细胞死亡方式[J].生物化学与生物物理进展,2013,40(2):137.
[3]陈鹏亮,郭进明,赖文杰,等.细胞程序性坏死的研究进展[J].华西医学,2016,31(8):1447.
[4]Fitzwalter BE,Thorburn A.Recent insights into cell death and autophagy[J].The FEBS journal ,2015,282(22):4279.
[5]Philipp S,Sosna J,Adam D.Cancer and necroptosis:friend or foe?[J].CMLS ,2016,73(11-12):2183.
[6]余永晟,李 金,黄 海,等.坏死性凋亡在恶性肿瘤中的研究进展[J].岭南现代临床外科,2016,16 (1):111.
[7]Hu X,Xuan Y.Bypassing cancer drug resistance by activating multiple death pathways-a proposal from the study of circumventing cancer drug resistance by induction of necroptosis[J].Cancer Lett,2008,259(2):127.
[8]毛庆祥,杨天德.胱氨酸/谷氨酸反向转运体的研究进展[J].生理科学进展,2014,45(6):434.
[9]Dixon S J,Lemberg K M,Lamprecht M R,et al.Ferroptosis:an iron-dependent form of nonapoptotic cell death[J].Cell,2012,149(5):1060.
[10]Lu B,Chen XB,Ying MD,et al.The role of ferroptosis in cancer development and treatment response[J].Front Pharmacol,2018,(8):992.
[11]Otsubo K,Nosaki K,Imamura CK,et al.Phase I study of salazosulfapyridine in combination with cisplatin and pemetrexed for advanced non-small-cell lung cancer[J].Cancer Sci,2017,108(9):1843.
[12]Su J,Liu F,Xia M,et al.p62 participates in the inhibition of NF-κB signaling and apoptosis induced by sulfasalazine in human glioma U251 cells [J].Oncol Rep,2015,34(1):235.
[13]Fulda S.Therapeutic exploitation of necroptosis for cancer therapy[J].Semin Cell Dev Biol,2014,35:51.
[14]Wang T,Jin Y,Yang W,et al.Necroptosis in cancer:An angel or a demon [J].Tumour Biol,2017,39(6):1.
[15]Zhang D W,Shao J,Lin J,et al.RIP3,an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis[J].Science,2009,325:332.
