基于信息共享的医用低温保存箱审评
2018-04-23张一夏文龙山东省食品药品监督管理局审评认证中心山东济南250014
张一 夏文龙 山东省食品药品监督管理局审评认证中心 (山东 济南 250014)
近年来,数据共享交换平台在社会各个领域的广泛应用促使信息共享共赢成为大数据时代科技发展的新趋势。作者以医用低温保存箱产品为示例,对基于信息共享的技术审评模式进行探讨,期望能为医疗器械技术审评人员进行技术审评提供参考。
1.基于信息共享的医疗器械审评模式介绍
基于信息共享的医疗器械技术审评模式,是指充分整合技术审评、行政审批、市场监管环节中的各类信息资源,将质量管理体系检查、临床试验检查、生产许可检查、监督检查、不良事件监测等信息都输入到技术审评环节,从而对产品的安全性有效性进行全面的评价。同时,也可以把技术审评环节信息反向输出到质量管理体系检查、监督检查等环节,制定个性化检查方案。消除技术审评、行政审批、市场监管等各个环节中的信息孤岛,实现对产品风险的整体把控,从而建立以审评为主导,检查检验等为支撑的技术审评体系。
2.医用低温保存箱技术审评过程中对多种信息的分析应用
2.1 对不良事件监测信息的分析应用
作者通过查询2015~2017年全国范围内医疗器械不良事件监测数据,发现医用低温保存箱产品在全国医疗机构使用过程中风险信号共出现41次。产品风险信号主要包括:使用过程中突然不制冷,温度失控;门体出现大量水珠;使用过程中不正常报警;压缩机声音明显增大;制冷剂泄露等。其中使用过程中突然不制冷,温度失控以及使用过程中不正常报警最常出现,其具体分布见图1。
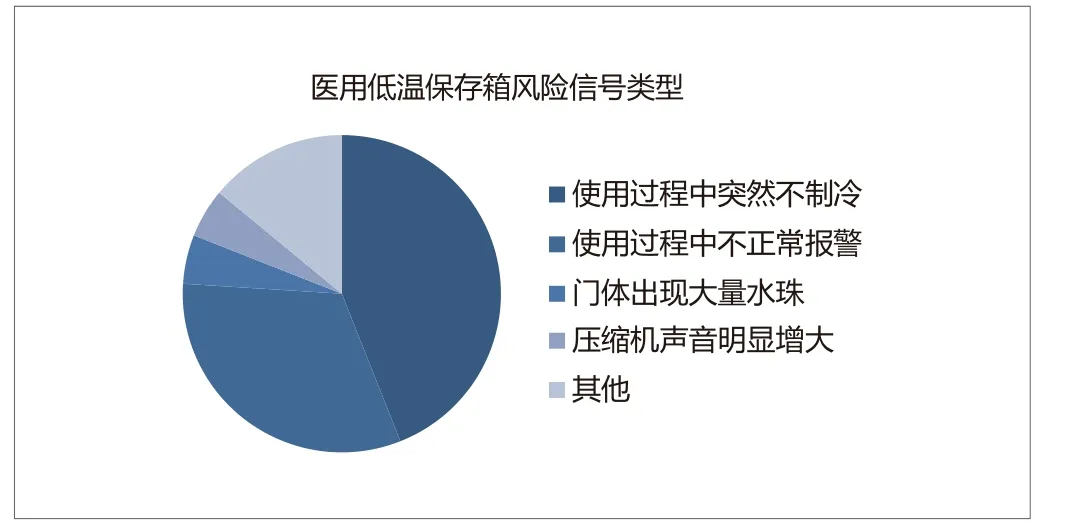
图1. 2015~2017年医用低温保存箱风险信号类型
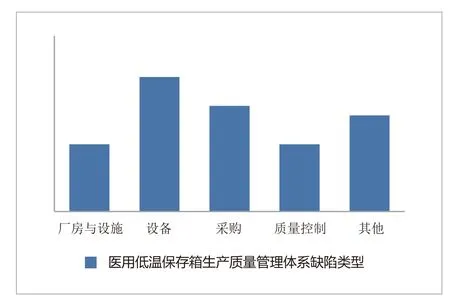
图2. 山东省2015~2017年度医用低温保存箱生产质量管理体系缺陷类型
对于上述风险信号分析其严重程度可发现,使用过程中突然不制冷,温度失控导致的储存物品失效,如血浆、成分血液、生殖细胞、器官等,其破坏性最高,往往造成比较严重的后果,不仅造成储存物品的浪费,对于可能预期进入人体的物品,存在较大的安全隐患。
而造成上述风险信号出现的原因主要包括:主板、控温器、电路质量不过关,使用时故障;箱门密封条老化;溢流管堵塞;风扇过滤网过脏;散热器设计位置不合理等。
故技术审评过程中,将上述风险信号特别是程度严重的风险信号以及风险信号产生的主要原因进行输入,在产品研究资料、产品技术要求、风险管理资料、使用说明书中进行重点关注。
2.2 对现场检查信息的分析应用
作者通过总结近2年来山东省对医用低温保存箱产品质量管理体系检查情况,发现质量管理体系常出现的问题主要包括:未按照待验、合格、不合格、退货或召回等进行有序、分区存放各类材料和产品;未配备与产品生产规模、品种、检验要求相适应的检验场所和设施;生产设备未有明显的状态标识;未建立供应商审核制度,未对供应商进行审核评价;采购纪录不齐全,未包括原材料清单、供应商资质证明文件、质量标准、检验报告及验收标准等;未对采购物品进行检验或验证;需要常规控制的进货检验、过程检验和成品检验项目不齐全等,其具体数量见图2。
虽然质量管理体系需要经整改合格,产品才会进入技术审评阶段,但仍可把发现的问题输入到技术审评阶段。因产品在原材料进货质量、进货检验以及过程控制等方面存在缺陷,故技术审评阶段可以对研究资料、生产制造信息、技术要求进行重点关注。
3.医用低温保存箱审评要点
统合上述所有信息,结合GB/T 20154-2014《低温保存箱》,形成了对手术无影灯产品的审评要点。
除根据“关于公布医疗器械注册申报资料要求和批准证明文件格式的公告”[1]对包括的13项资料进行逐项审评外,需要对上述的研究资料、生产制造信息、产品技术要求、风险管理资料以及使用说明书中提到的信息进行重点关注,以确保在技术审评阶段将产品风险控制在可以接受的水平。
在研究资料中,应关注制冷原理类型、制冷剂类型,对其原理以及设计的安全性、有效性进行审评,如其散热器设计在下部,不利于散热,则在使用过程中有温度过高的风险;还应关注产品可储藏物品类型及必要的证据,如可用于对生殖细胞的储存,则需验证降温速度等对其的损伤,否则复温使用过程中复活率会大大下降;同时,应关注产品使用寿命的研究,若采用关键元器件老化试验验证其使用寿命,则应对关键元器件的选择以及老化试验方法选择的合理性说明其依据,而对于可重复使用的组件,应当验证其使用次数,对其使用寿命、使用频率的合理性进行审评。因生产质量管理存在一定缺陷,故应关注相应验证过程是否在完善的质量管理体系下进行,同时验证过程的完整性及适宜性。
在生产制造信息中,应重点关注工艺流程及其过程控制,并识别关键过程和特殊过程,且明确研制、生产场地的制造和检验等具体情况。工艺流程一般需要包括原材料收货、来料检验、原材料入库、原材料出库、发泡、焊接、抽空、灌注、检漏、产品测试检验等过程,而对其中的特殊过程,应进行确认。
在产品技术要求中,应按照GB/T 20154-2014《低温保存箱》关注特性点温度、温度均匀性、降温时间等关键技术指标,还应重点关注结构和材料性能。而对产品软件功能以及逐步出现的联网功能、远程监控功能都应进行验证。
在风险管理资料中,应关注是否对囊括上述风险信号的所有危险(源)进行了识别,是否列明可预见的事件、危险情况以及可发生的伤害;还应关注其风险控制的方案是否合理、实施方法是否严格遵循方案设计以及风险控制的完整性和适宜性。
在使用说明书中,应关注是否清晰且详细的对如何进行医用低温保存箱的调试、操作、使用、清洁消毒、维护保养等进行说明,并易于理解,还应对上述过程提供相应培训。而因制造商在对适用范围内所包含物品进行冷冻效果验证时不可能涵盖临床层面所有可能因素,故在说明书中应提示使用者在使用前对冷冻效果进行相应验证,并对其冷冻效果进行实时监测。
4.小结
医疗器械技术审评是建立在具有充分信息基础上的一种决策[2]。故除了上述的不良事件监测信息以及质量管理体系检查信息外,技术审评阶段还可输入检验信息、临床试验检查信息、监督检查信息等,如检验过程中常出现的不合格项、临床试验检查中存在的主要问题、监督检查中常出现的缺陷项等,对这些信息进行分析并在技术审评阶段提出解决方案。通过整合以上所有的信息来形成对于该类产品的基于风险的审评要点,从而实现对产品风险的整体把控。同时,还可以把技术审评阶段整合的信息反向输出到现场检查等阶段,制定该类产品的个性化现场检查方案,或是反向输入到不良事件监测阶段,对该类产品的相关风险点进行重点监测与分析,从而建立以审评为主导,检查检验等为支撑的技术审评体系。而通过基于信息共享的医疗器械技术审评模式,势必对深化医疗器械审评制度改革,提高审评一致性起到推动作用。
[1] 国家食品药品监督管理总局.医疗器械注册申报资料要求和批准证明文件格式 [EB/OL].2014-09-05.
[2] 范乙.信息技术对我国药品技术审评工作的保障作用[J].中国药事,2009,23(8):752-754.
