汽油典型烃分子氧化生胶机理的分子模拟
2018-04-12陶志平代振宇
李 娜, 龙 军, 赵 毅, 陶志平, 代振宇
(中国石化 石油化工科学研究院, 北京 100083)
汽油从油箱经油路系统输送至喷嘴和进气阀的使用中,在发动机内高温和氧气存在的情况下,汽油中不稳定烃分子较易发生氧化缩合等一系列反应生成相对分子质量较大且极性较高的化合物[1-3],生成的大分子化合物在极性较低的汽油中油溶性较差,通过分子中的极性基团与金属表面产生分子间相互作用进而吸附在金属表面。发动机经过长期运转,容易在喷嘴、进气阀及燃烧室等部位形成大量沉积物[4]。沉积物的产生会导致油耗增加、发动机动力损失、点火不良、操作不灵等问题,严重影响发动机的工作效率及尾气排放[5-6]。
通常认为,汽油在发动机内生成沉积物的实质是链式自由基反应[7-8],包括链引发、链增长和链终止:
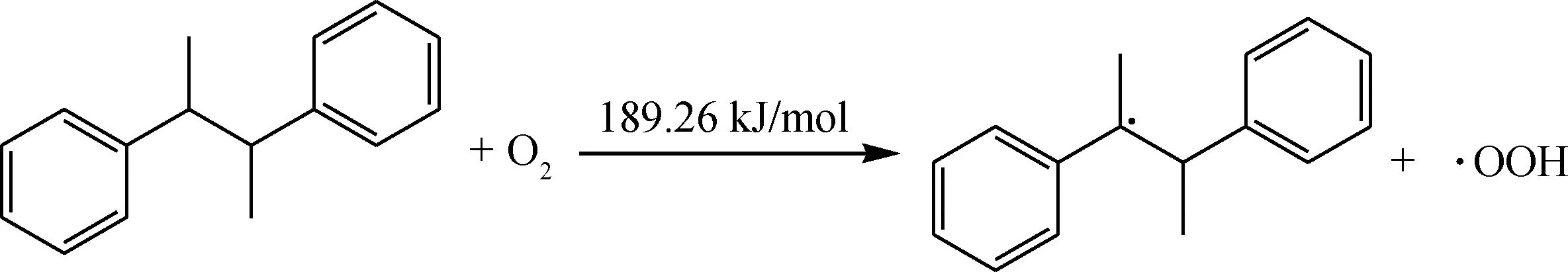
(1)链引发:RH+O2→R·+HOO·
(2)链增长:R·+O2→ROO·
ROO·+RH→ROOH+R·
ROOH→RO·+·OH
RO·+RH→ROH+R·
·OH+RH→H2O+R·
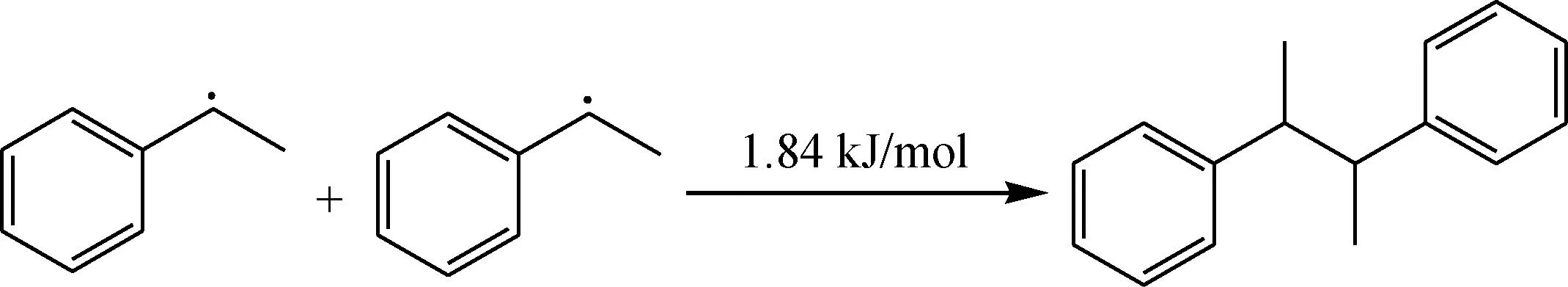
(3)链终止:R·+·R→R—R
ROO·+ROO·→ROOOOR→
R′C=O+ROH+O2
目前文献中大多采用实验手段来研究汽油烃分子氧化过程中可能产生的中间产物,从而推测生成胶质的可能反应路径。Story等[9]认为,催化裂化汽油暴露在空气中生成的胶质是有机过氧化物、醛、酮、酸的混合物,推测胶质是以过氧化物的形成作为引发步骤的氧化反应的产物,而且与过氧化物的氧化分解紧密相关[10]。Tseregounis等[11]利用氧化的燃料研究沉积物的形成过程发现,汽油中的极性预氧化产物是产生沉积物的前驱体,这些油溶性的前驱物会进一步形成非油溶性物质,最终附着在金属表面形成沉积物。Batts等[12]认为,中性化合物氧化形成极性的中间产物可能是形成沉积物的主要路径,胶质的生成与两个过氧自由基结合的链终止反应有关[13]。近年来,Pfaendtner等[14-15]结合机理自动生成技术(Automated mechanism-generation)和量子化学理论方法,系统地研究了癸烷分子的氧化反应网络,他们将反应过程中可能涉及到的数千种反应缩减为占主导作用的典型反应,从而建立了全面且准确的氧化反应网络。
然而,由于汽油烃分子氧化生胶过程十分复杂,现有文献报道仅仅是通过实验手段对可能发生的化学反应以及可能生成的产物进行了研究,相对孤立而零散。其中,涉及到的关键基元反应更是因为实验手段无法捕捉自由基而无法揭示微观反应。日臻成熟的分子模拟技术,为从分子和原子水平上认识汽油烃分子的氧化生胶反应历程提供了可能。从基元反应角度入手,借助分子模拟手段对其关键反应步骤的难易程度进行定量分析,有利于更充分地认识烃分子氧化生胶过程中的关键基元反应和关键活性中间体,进而抑制其生胶。
1 研究汽油氧化生胶机理采用的模型化合物和方法
1.1 汽油分子模型化合物的选择
汽油为C4~C12的烷烃、环烷烃、异构烷烃、烯烃和芳香烃等的复杂混合物。影响汽油氧化生胶的最根本原因是其化学组成。一般认为,汽油中的烷烃、环烷烃和芳烃在常温下均不易发生氧化反应,而其中的各种不饱和烃,特别是环烯烃和二烯烃容易发生氧化缩合等反应,从而生成胶质[16-17]。但是由于目前汽油质量升级,成品汽油中基本不含二烯烃和环烯烃,因此这里选取汽油中常见的单烯烃作为烯烃的模型化合物。笔者选取碳数为8的不同类型汽油烃分子作为模型化合物,其分子结构如图1所示,数字编号代表C—H键的不同位置。
1.2 计算方法
研究汽油烃分子氧化结胶反应过程,关键在于搜索基元反应的过渡态,计算反应的能量势垒(简称能垒)。由量子化学方法获得的反应能垒,是0 K时过渡态能量与反应物能量的差值,即反应物转化为生成物过程中跨越能量最高时所需要的能量。
采用DaussaultBiovia公司的分子模拟软件Materials Studio 8.0中的DMol3量子力学模块进行基于密度泛函方法的相关理论研究。计算过程中选用基于广义梯度近似(GGA)的PBE泛函[18]、DNP基组,使用自旋极化方案处理开壳层体系,自洽场(SCF)迭代收敛的阈值设为2.63×10-2kJ/mol。收敛精度为:能量5.25×10-2kJ/mol,受力10.50(kJ·mol-1)/nm,位移5×10-4nm。过渡态搜索采用完全线性同步和二次同步变化(Complete LST/QST)方法。搜索出的过渡态处于势能面的“鞍点”处,是连接反应物和产物的最低能量路径上的极大值点,同时是其他方向上的极小值点。过渡态结构的能量二阶导数的本征值仅有1个负值,即过渡态有且仅有1个虚频。
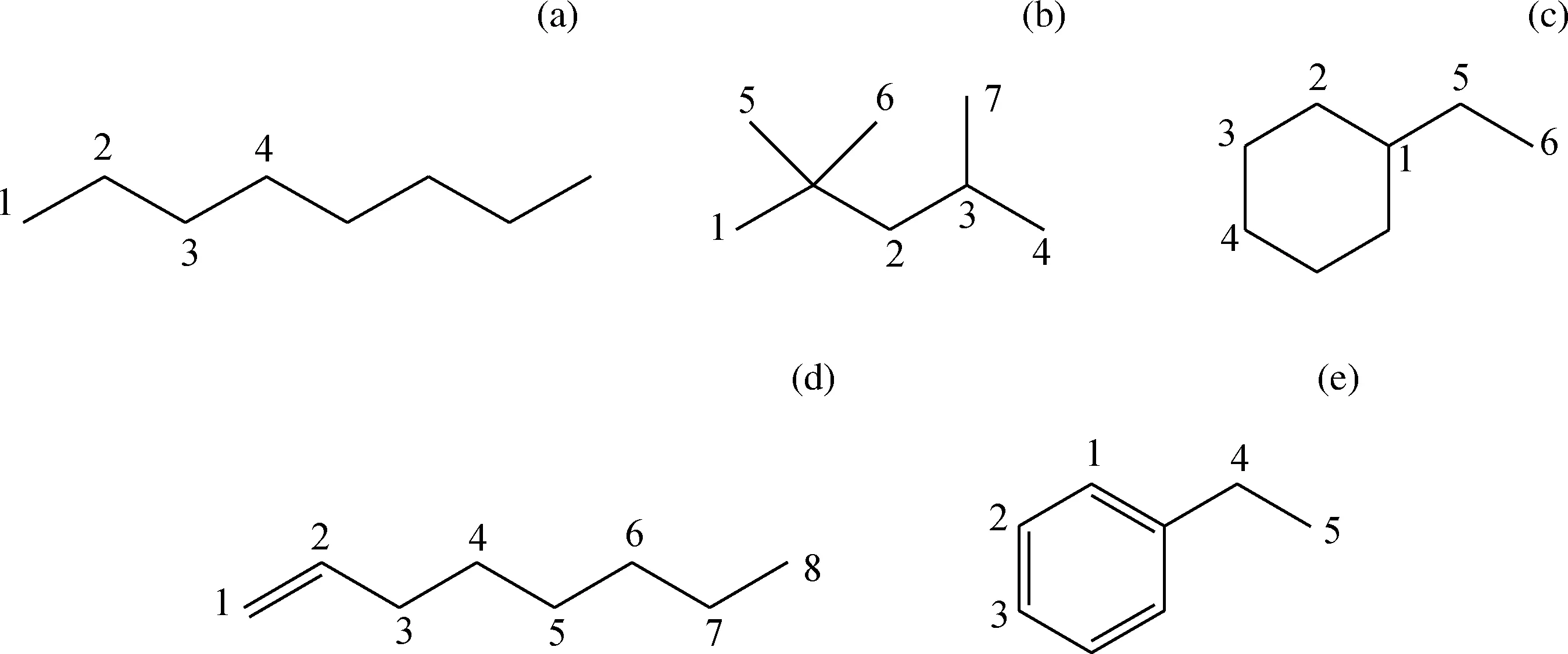
图1 汽油分子模型化合物结构Fig.1 The molecular structure of gasoline model compounds(a) Octane; (b) Isooctane; (c) Ethylcyclohexane; (d) 1-Octene; (e) Ethylbenzene
2 结果与讨论
2.1 不同汽油烃分子C—H键均裂能对比
依据烃分子氧化的链式自由基反应机理,烃分子的C—H键可能被氧分子进攻,从而引发自由基的生成,同时烃分子主要与活性自由基发生氢转移反应进而不断生成新自由基[19]。因此,C—H键是烃分子氧化的反应活性位置。表1为模型化合物(a)~(e)不同位置C—H键的均裂能数据。由不同位置C—H键的均裂能数据可以看出,相比于伯碳位置C—H键和仲碳位置C—H键,叔碳位置C—H键的均裂能较低;对于含有烯烃双键或苯环的分子结构,α位C—H键的均裂能最低。相应的5种模型化合物被氧分子进攻的C—H活性位分别为C2—H、C3—H,C1—H、C3—H、C4—H,从而为后续链引发反应的计算提供了进攻活性位。
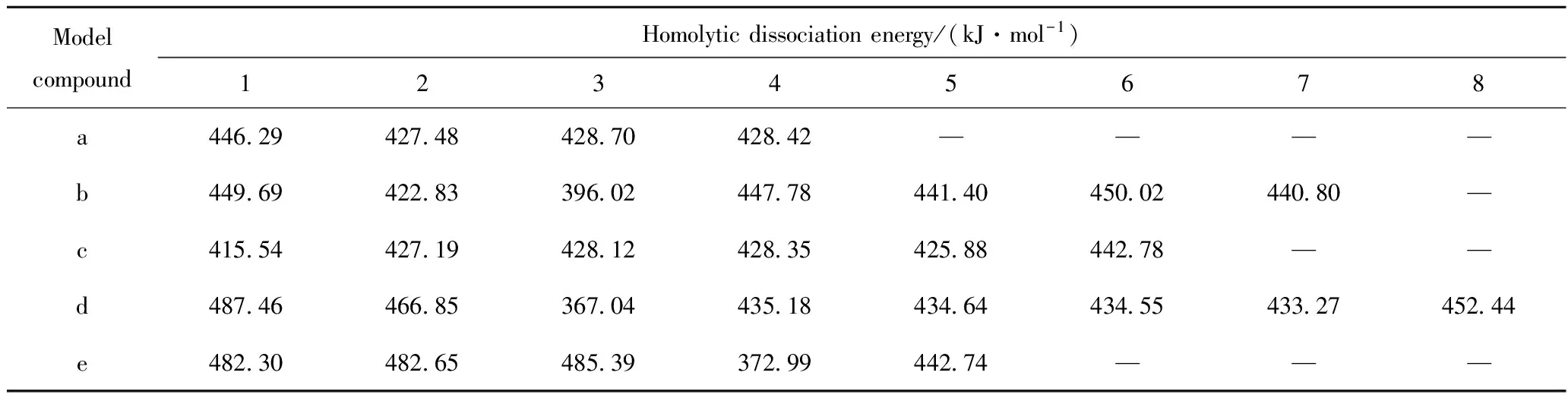
表1 模型化合物(a)~(e)不同位置C—H键均裂能对比Table 1 Homolytic dissociation energies Comparison of C—H bond in model compounds(a)-(e)
2.2 氧化生胶机理
2.2.1正辛烷的氧化生胶机理
对于正辛烷,活性位为C2—H,首先O2分子进攻正辛烷的活性C—H位,夺取氢原子,产生第1个自由基,从而引发链式自由基反应,链引发:
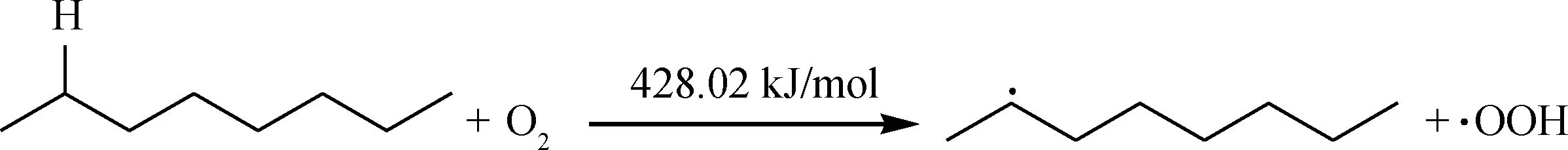
然而,该引发反应的能垒较高,在较低温度环境下很难发生,因此,笔者认为正辛烷氧化安定性较好,不参与汽油的氧化自由基反应,进而不会贡献生成沉积物。
2.2.2异辛烷的氧化生胶机理
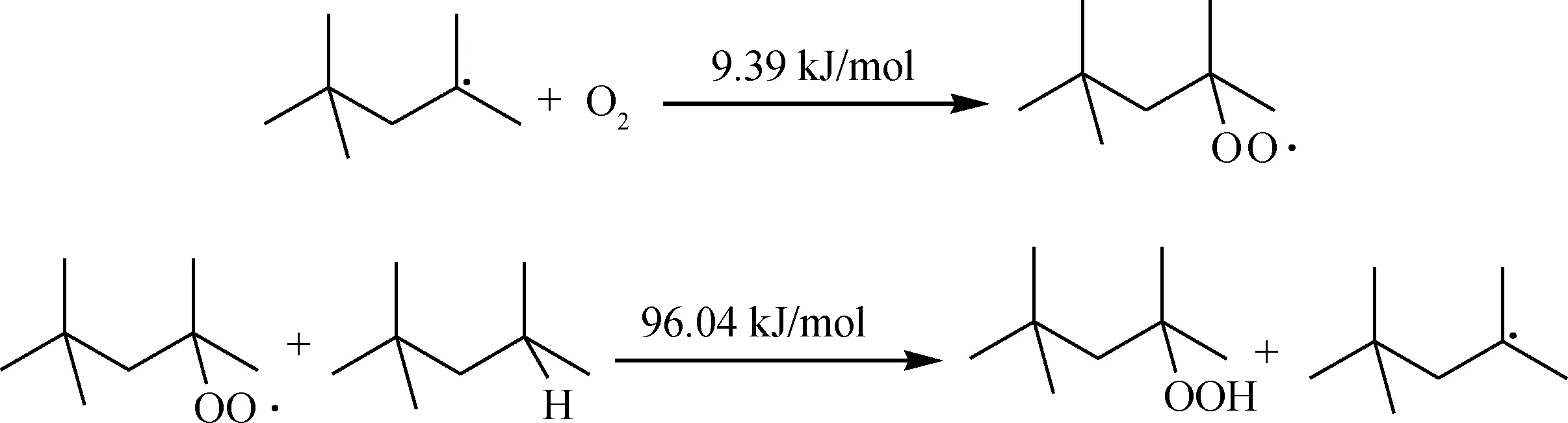
对于异辛烷,其最弱的C—H键为叔碳位置C3—H,由于叔碳连接的3个甲基叠加的吸电子能力较强,使得连接的C—H键均裂能较低,因此O2分子首先进攻该活性位,引发链式自由基反应,由反应能垒数据可知,该引发反应需要的能垒较低,在较低温度下较易发生。
链引发:
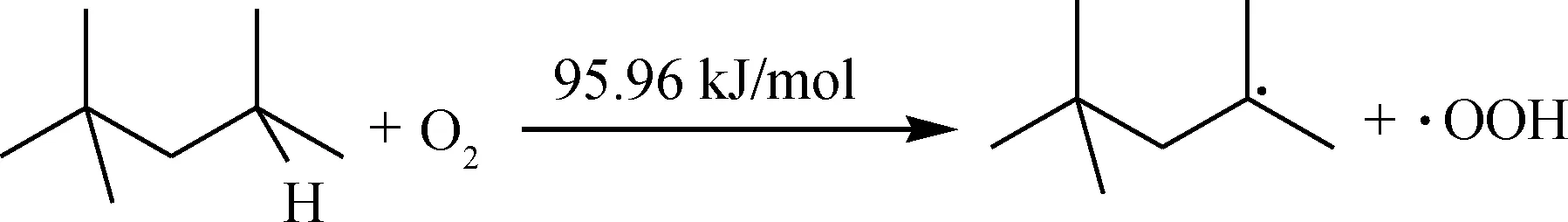
生成的烃分子自由基会迅速被氧气进攻,结合生成过氧自由基,该类反应能垒非常低;生成的过氧自由基会进攻其他新的烃分子,生成更多的自由基参与链反应,将反应链继续传递,同时生成过氧化物。
链传递:
生成的过氧化物会进一步发生均裂反应生成烷氧自由基和羟基,这两种自由基反应活性较高,会迅速进攻新的烃分子夺取氢原子,生成更多的自由基参与反应,加剧链式反应的进行。
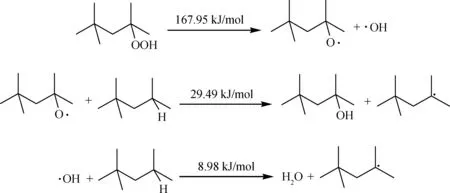
生成较多自由基后,自由基之间极易发生结合生成较为稳定的化合物,该反应产物相对分子质量明显增大,是贡献生成沉积物的反应方向。
链终止:
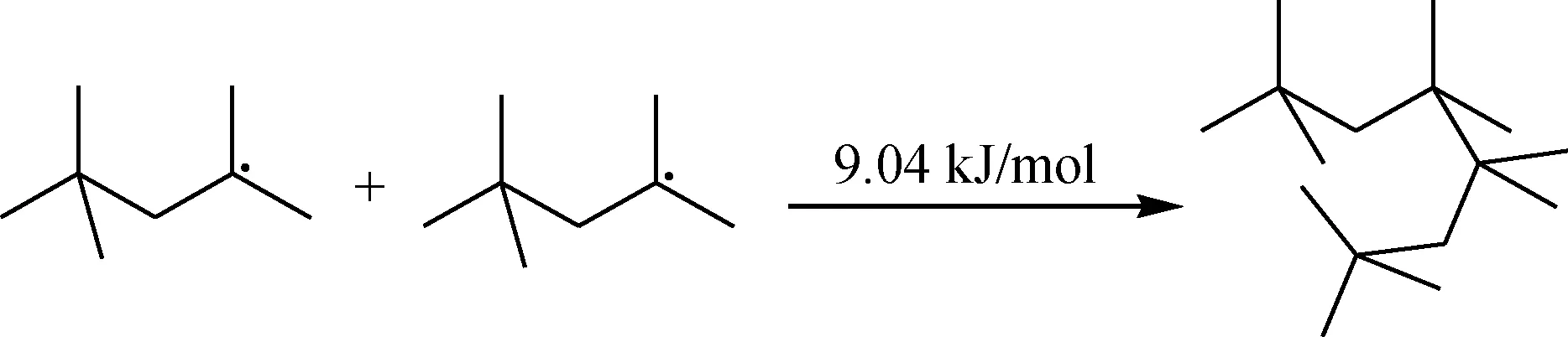
生成较为稳定的化合物之后,O2分子继续进攻均裂能较低的仲C—H键,二次氧化(链引发):
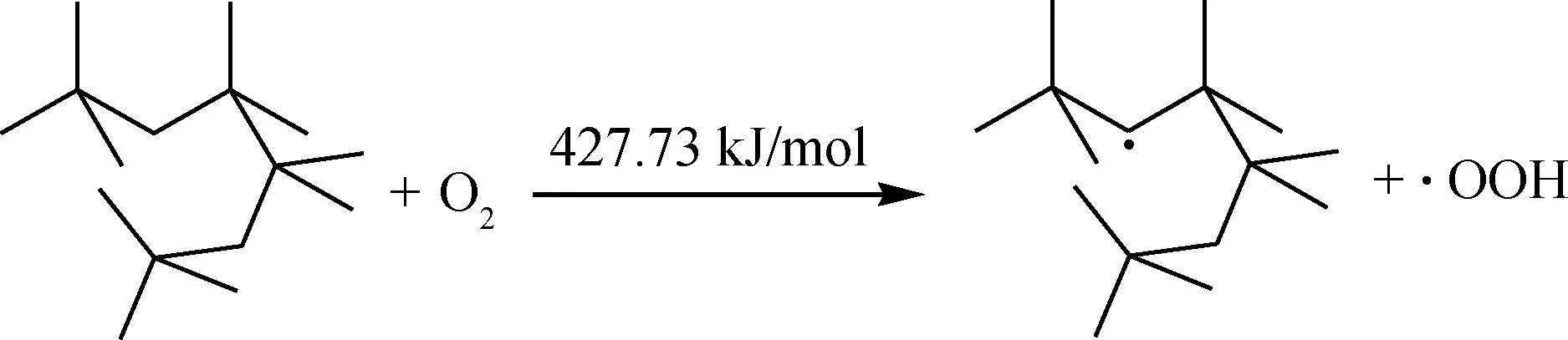
由此可知,该反应能垒非常高,在温度较低的情况下,无法跨越如此高的能垒,可以认为该反应无法进行。
由以上反应网络分析可知,异辛烷一次氧化的链终止反应的反应能垒非常低,可以认为异辛烷的氧化链式自由基反应极易发生链终止反应,从而泯灭自由基生成较为稳定的产物;同时该产物难以继续被二次氧化,因此可以认为,异辛烷虽然比较容易被氧气进攻引发链式自由基反应,但是深度氧化的可能性较小,因此异辛烷是清净性较好的组分,不是贡献生成沉积物的前驱体。
2.2.3乙基环己烷的氧化生胶机理
对于乙基环己烷,其最弱的C—H键为叔碳位置C1—H,叔碳连接的3个甲基叠加的吸引电子能力较强,使得相应的C—H键均裂能较低,因此O2分子首先进攻该活性位,夺取氢原子引发链式自由基反应。
链引发:
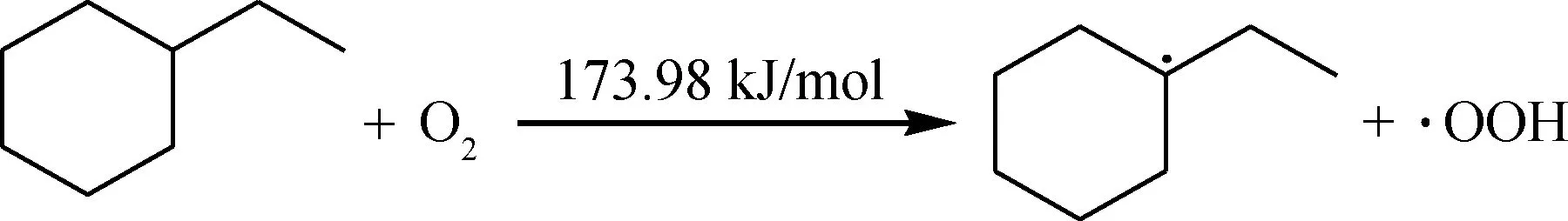
由反应能垒数据可知,该引发反应需要的能垒较低,在较低温度下可以发生。生成的自由基极易被O2进一步进攻,结合,生成过氧自由基,进而夺取其他烃分子的氢,生成更多的自由基参与链反应。
链传递:
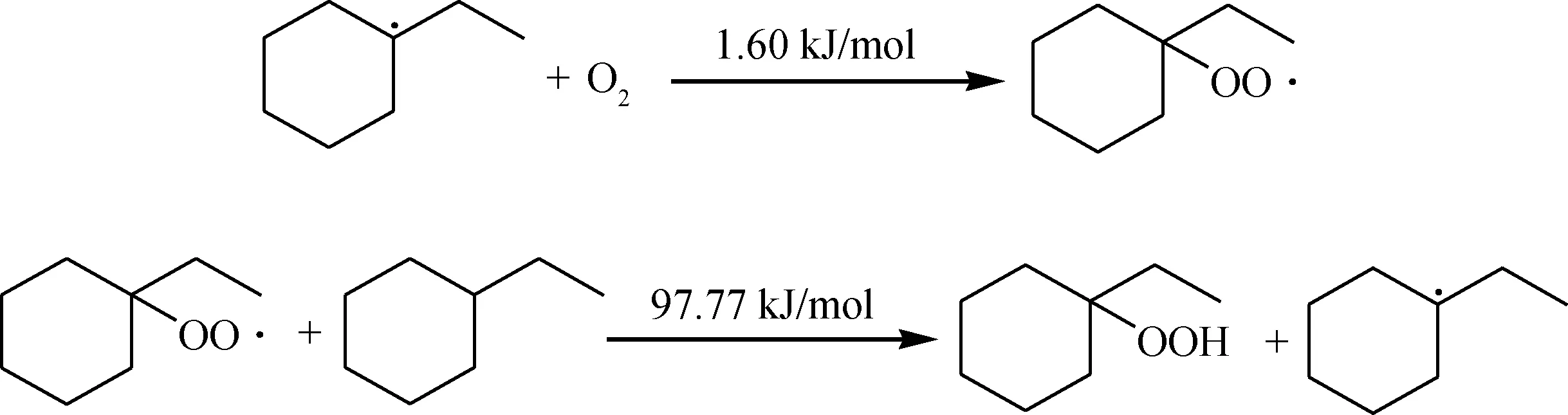
生成的氢过氧化物会进一步裂解生成活性较高的烷氧基和羟基,进而继续夺取烃分子氢原子生成更多的烃自由基参与链反应。
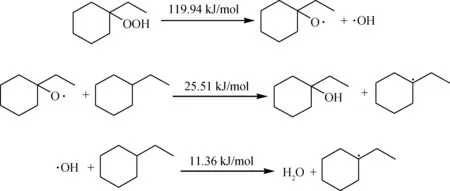
生成较多的烃自由基后,会发生自由基之间的偶联反应,生成较为稳定的产物。
链终止:
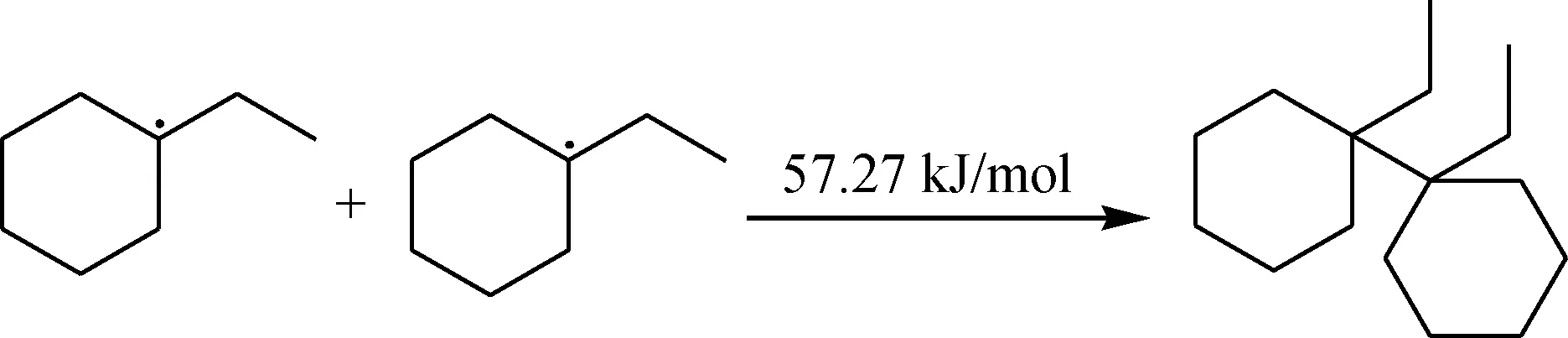
生成的较为稳定的产物会进一步被O2进攻较弱的C—H键,通过计算各个C—H位的键能,按照如下方式进行进一步氧化反应。可以看出该反应能垒非常高,因此,可以认为该反应在较低温度环境下无法进行。
二次氧化(链引发):
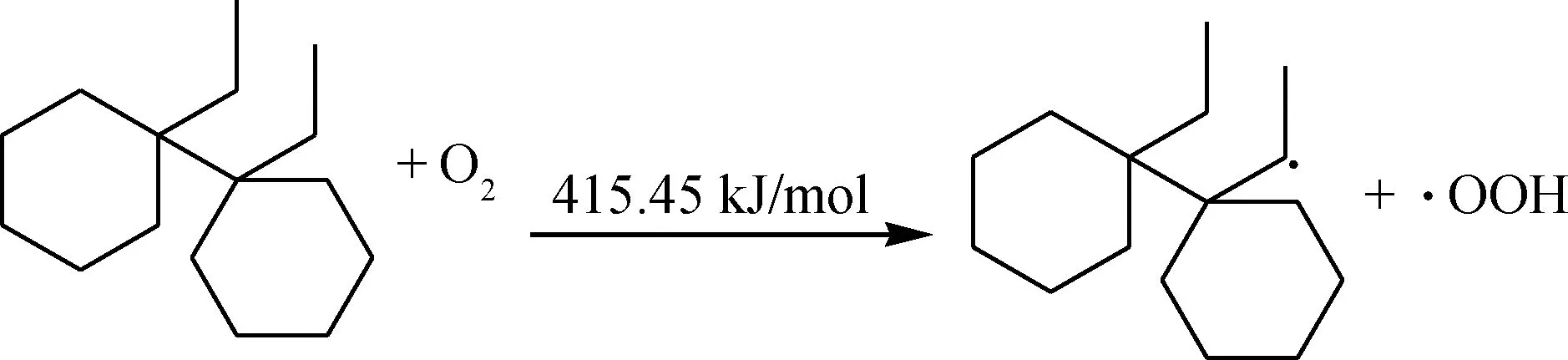
由此可见,乙基环己烷发生一次氧化反应的链终止反应能垒较低,可以认为乙基环己烷的氧化链
式自由基反应较易发生链终止反应,泯灭自由基生成较为稳定的产物,同时该产物难以继续被氧化,因此可以认为乙基环己烷不会贡献生成相对分子质量大且含氧极性较强的沉积物。
2.2.41-辛烯的氧化生胶机理
对于1-辛烯,其最弱的C—H键为烯烃双键α位置的C3—H,双键中的π键具有较强的吸电子能力,使得连接的C—H键的均裂能较低,因此O2分子首先进攻该活性位,夺取氢原子引发链式自由基反应,由反应能垒数据可知,该引发反应需要的能垒较低,在较低温度下可以发生。
链引发:
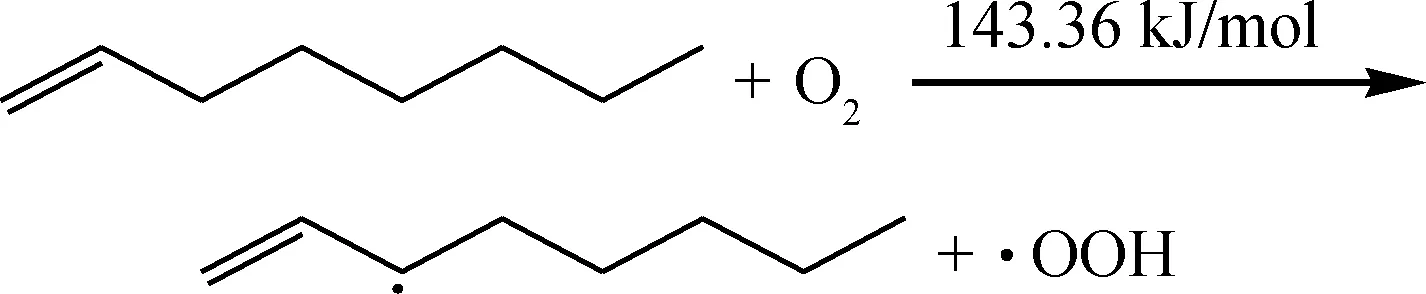
自由基一旦产生便可发生进一步的链传递反应。烃类自由基与氧气分子的加成反应很快,反应能垒接近0,生成的过氧自由基继续进攻新的烃分子产生更多的自由基参与链式反应,将反应传递下去。
链传递:
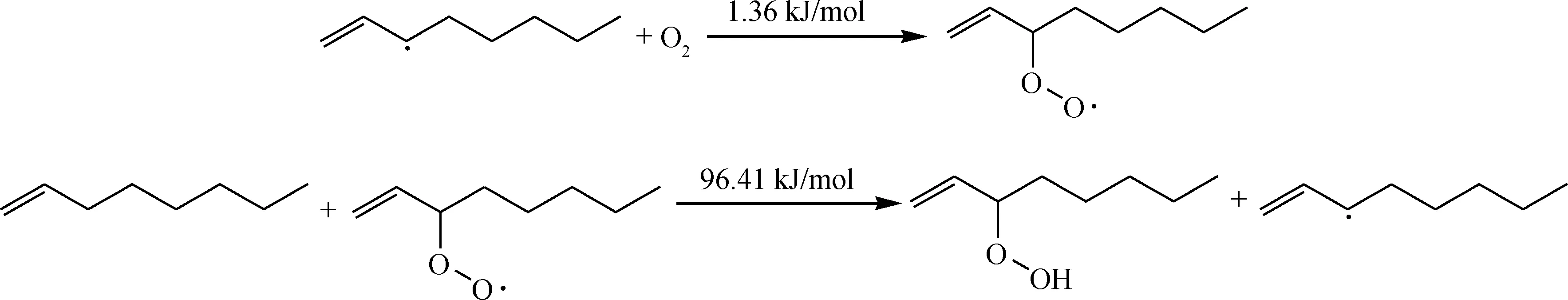
氢过氧化物ROOH中O—O键断裂,裂解为2个活性较高的自由基:烷氧基和羟基。
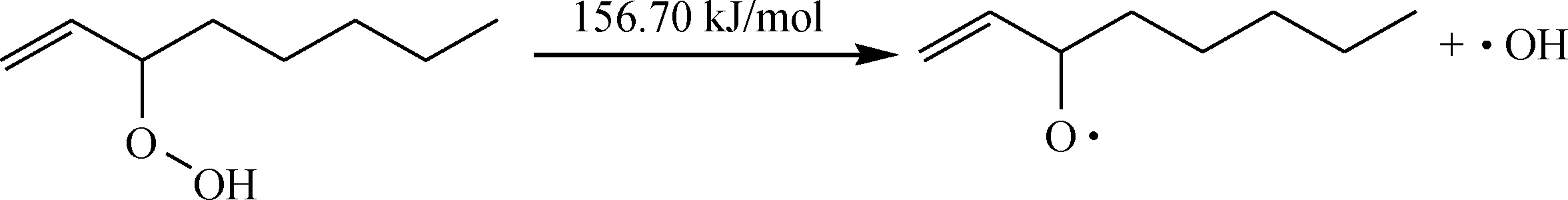
生成的烷氧基和羟基可以继续夺取烃分子的氢,产生更多的自由基,又可引发新的链反应:
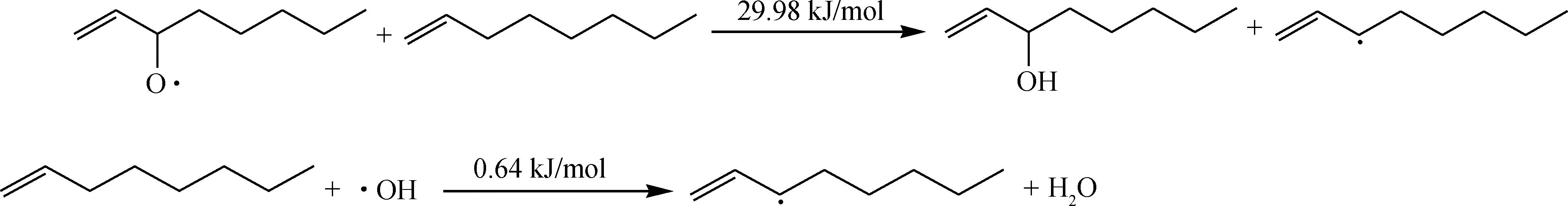
烷氧基和羟基进攻新的烃分子的反应能垒非常低,可见,这2个自由基反应活性非常高,一旦生成就难以控制其进一步进攻新的烃分子。
当R·和ROO·积累到一定程度时,极易发生终止反应,生成较为稳定的化合物。自由基的消失主要由于它们之间的相互结合,此外也可能发生在和器壁的碰撞中。这类反应的能垒较低,其中,R·+·R→R-R 的反应能垒更低,并且为相对分子质量增大的反应方向,二次氧化反应在此基础上继续进行。
链终止:
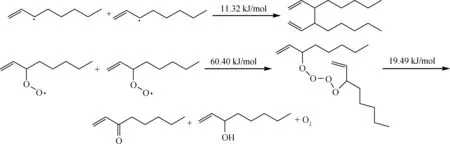
通过建立1-辛烯一次氧化反应网络发现,1-辛烯分子一旦引发生成烯烃自由基,与O2反应生成过氧自由基的能垒近乎为0,这表明,生成烯烃过氧自由基的反应将不可避免且无法有效抑制。过氧自由基继续进攻新的烃分子,反应能垒较低。这表明,生成的过氧自由基容易进攻烃分子夺氢,导致生成新的烃自由基,进而形成自由基链反应的不断循环。生成的氢过氧化物存在较弱的O—O键,其均裂的能垒为156.70 kJ/mol,而均裂反应后会生成2个含氧自由基:烷氧基和羟基。这2个自由基与新的1-辛烯分子反应的能垒非常低,低于过氧自由基的夺氢反应,表明烷氧基和羟基是2个非常活泼的自由基,一旦生成,会迅速从1-辛烯分子中夺氢,进而引发2个新的自由基链反应,导致链反应支链化。
生成的相对分子质量较大的产物会继续被氧气进攻,引发二次链式自由基反应。氧气分子进攻双键α位C—H,双键中π键的吸电子作用使得该位置的C—H键较弱,反应较易发生。
二次氧化(链引发):
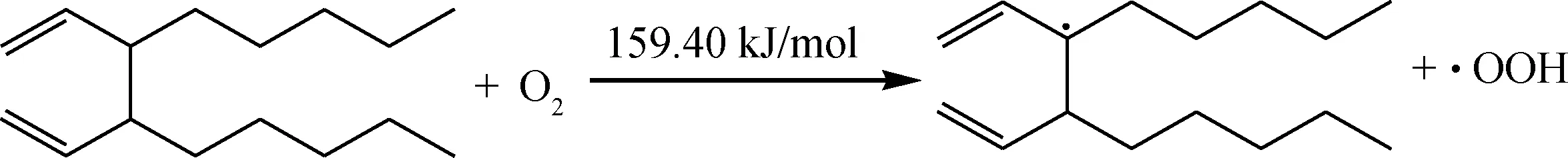
一旦引发第2次链式自由基反应,同样会迅速进一步发生链传递反应。二次氧化反应网络见图2和图3,蓝色虚框标识为该反应网络的反应物和产物。反应网络中主要发生脱氢、环化、加氧等反应,反应向相对分子质量增大、极性增强的方向进行,生成沉积物的反应网络始于链引发反应,止于链终止过程,最终产生稳定产物。
结合一次氧化反应历程以及二次氧化反应网络,可以更加直观地看出,反应能够进行取决于上一步反应产物中存在双键α位C—H键,或者存在C=O极性官能团,该含氧基团和双键中的π键吸引电子能力较强,能够降低该C—H键的电子云密度,进而降低其均裂能,因此使得氧化反应能够继续进行。
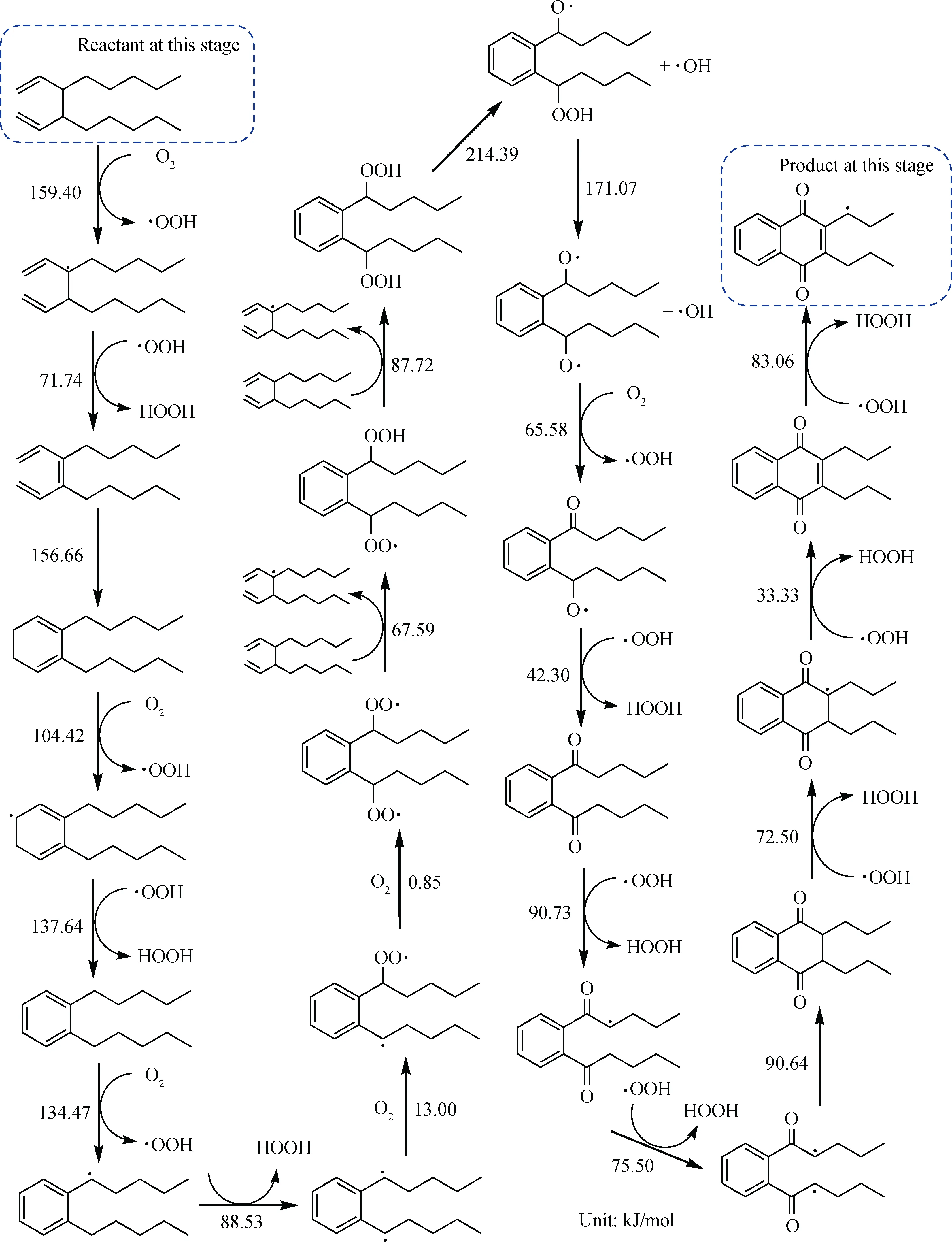
图2 1-辛烯二次氧化反应网络(1)Fig.2 The second oxidation network(1)of 1-octene
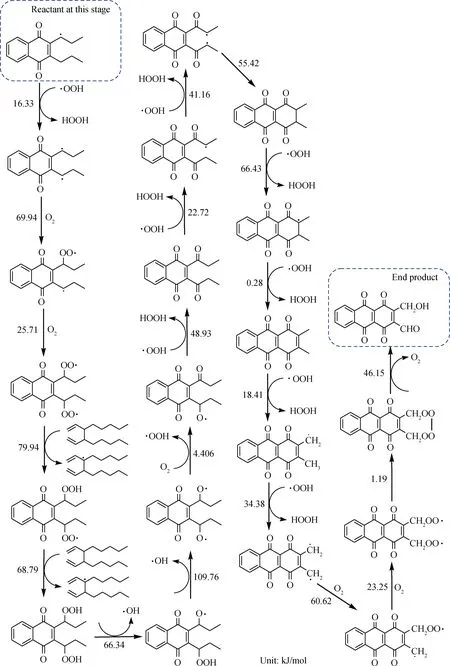
图3 1-辛烯二次氧化反应网络(2)Fig.3 The second oxidation network(2)of 1-octene
在自由基链反应过程中,过氧自由基进攻新的烃分子夺氢的反应以及氢过氧化物O—O键的均裂反应能垒相对较高,是决定1-辛烯氧化快慢的控速步骤,从这两个反应出发,清除过氧自由基和氢过氧化物是抑制该氧化生胶过程的关键措施。
2.2.5乙苯的氧化生胶机理
首先O2分子进攻乙苯较弱的苯环α位的C—H键,夺取氢原子,生成自由基,引发链式反应。
链引发:
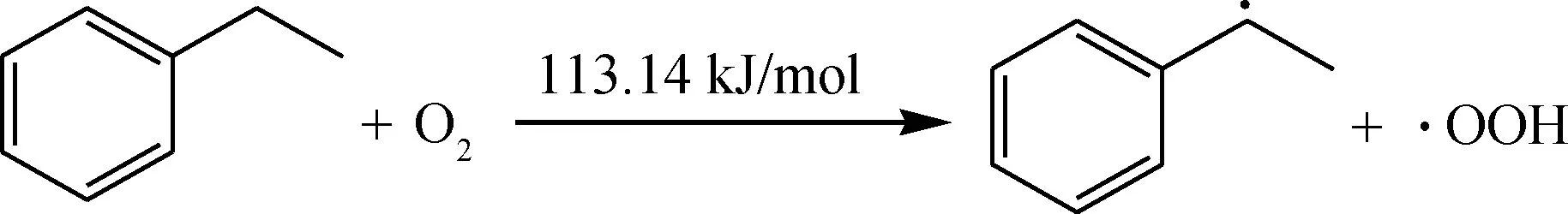
生成自由基后会迅速被氧气进攻生成过氧自由基,过氧自由基进一步夺取烃分子的氢,生成更多的自由基参与链反应。
链传递:
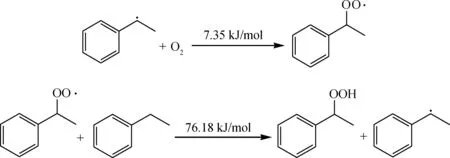
生成的过氧化物会进一步裂解为2个活性较高的自由基,烷氧基和羟基,它们极易夺取烃分子的氢,生成更多的烃自由基参与链反应。
烃自由基达到一定浓度后会,自由基之间会发生偶联反应,生成较为稳定的产物,反应能垒非常低。
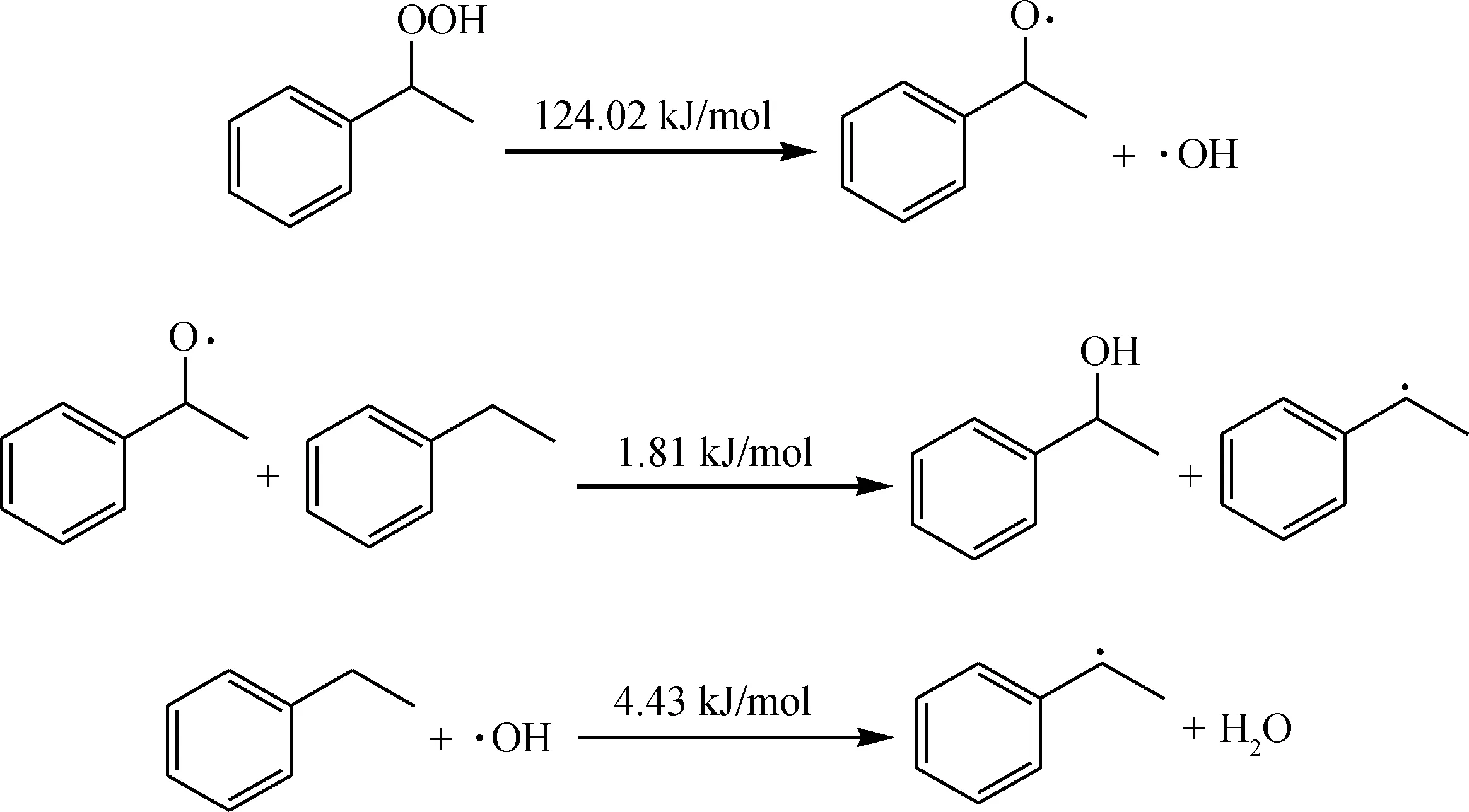
生成的产物会继续被O2分子进攻引发新一轮的氧化反应,该反应能垒不是特别高,可以在低温下发生。
二次氧化(链引发):
生成自由基后会迅速被O2进攻生成过氧自由基,进而夺取其他烃分子的氢,产生更多自由基。二次氧化反应网络如图4所示,主要发生脱氢、加氧、成环反应,生成二醚结构后进一步被O2夺氢,反应能垒非常高,可以认为不能继续发生,因此该二醚结构为最终反应产物。与文献[20]中报道的芳烃分子氧化缩合生成的大分子极性化合物特点相符。因此,乙苯是贡献生成沉积物的前驱体。
通过分析该反应网络,其反应能够一直进行取决于上一步生成的产物中存在苯基α位C—H、叔位C—H或者双键α位C—H。因为苯基中的大π键、3个甲基的σ键或双键中的π键均具有较强的吸引电子的能力,能够降低相邻C—H键的电子云密度,从而降低其均裂能,因此,进行氧化脱氢的反应能垒较低,反应能够进一步进行。
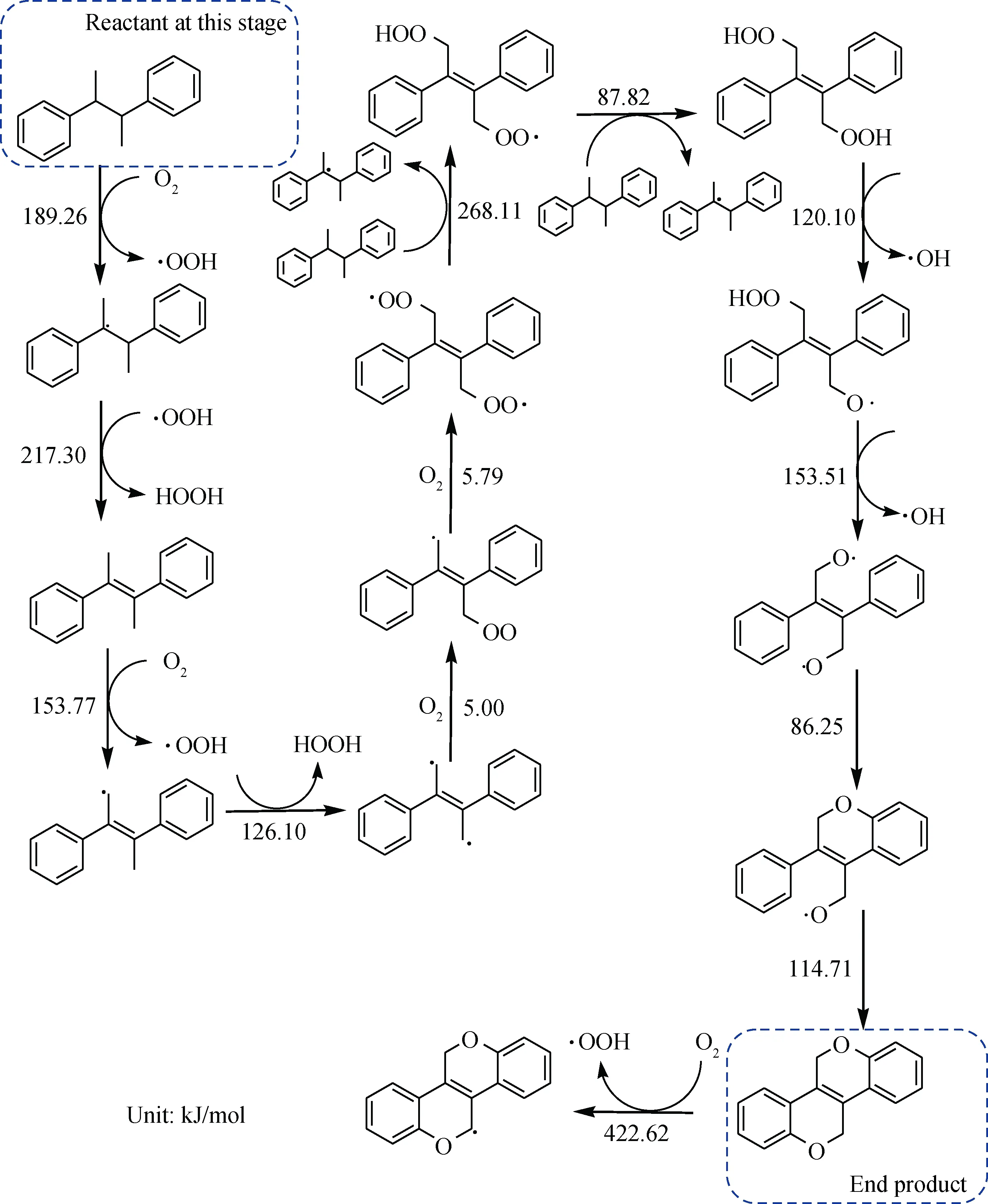
图4 乙苯二次氧化反应网络Fig.4 The second oxidation network of ethylbenzene
3 结 论
依据烃分子氧化的链式自由基反应机理,对汽油典型烃分子氧化生胶的反应网络进行了分子模拟研究,得出以下结论:
(1)沉积物是汽油中烃分子通过自由基氧化、脱氢、自由基之间缩合、环化等过程逐渐形成的;反应方向为相对分子质量增大、极性增高。
(2) 烃分子的结构特点决定了是否会与O2通过链式自由基反应贡献生成相对分子质量较大、极性较高的沉积物。
(3)烃分子结构中存在双键α位C—H、叔位C—H、苯基α位C—H容易引发链式自由基反应,但能否继续向沉积物的反应方向进行,取决于相对分子质量增大后的产物中是否存在双键α位C—H、叔位C—H、苯基α位C—H或者C=O这种使得相邻C—H键变弱的吸电子基团,容易被继续夺氢将反应继续传递下去。
(4)氧化生胶反应过程中,过氧自由基ROO·和烃过氧化物ROOH是最关键的2个中间体,ROO·与烃分子的反应,以及ROOH的O—O键均裂反应,是决定氧化快慢的速控步骤。抑制汽油烃分子氧化的关键措施在于清除ROO·以抑制其从烃分子上夺氢的反应;清除ROOH以抑制其O—O键均裂直接生成RO·和·OH 2个活性非常高的自由基的反应。
[1] BROOKS B T. The Chemistry of gasolines-particularly with respect to gum formation and discoloration[J].Industrial and Engineering Chemistry, 1926, 18(11): 1198-1203.
[2] FLOOD D T, HLADKY J W, EDGAR G. Chemical nature of gum-forming constituents in gasoline[J].Industrial and Engineering Chemistry, 1933, 25(11): 1234-1239.
[3] WALTERS E L, MINOR H B, YABROFF D L. Chemistry of gum formation in cracked gasoline[J].Industrial and Engineering Chemistry, 1949, 41(8): 1723-1729.
[4] NAGPAL J M, JOSHI G C, SINGH J. Gum forming olefinic precursors in motor gasoline: A model compound study[J].Fuel Sci Technol Int, 1994, 12(6): 873-894.
[5] LACEY P, GAIL S, KIENTZ J M, et al. Internal fuel injector deposits[J].SAE Int J Fuels Lubr, 2012, 5(1): 132-145.
[6] TANAKA A, YAMADA K, OMORI T, et al. Inner diesel injector deposit formation mechanism[J].SAE Int J Fuels Lubr, 2013, 6(3): 755-761.
[7] PRICE R J, WILKINSON J T, JONES D J, et al. A laboratory simulation and mechanism for the fuel dependence of SI combustion chamber deposit formation[C]//Toronto: 1995-10-16-19, SAE paper, 952445.
[8] DIABY M, SABLIER M, LENEGRATE A, et al. Understanding carbonaceous deposit formation resulting from engine oil degradation[J].Carbon 2009, 47(2): 355-366.
[9] STORY L G, PROVINE R W, BENNETT H T. Chemistry of gum formation by cracked gasoline[J].Industrial and Engineering Chemistry, 1929, 21(11): 1070-1084.
[10] MORRELL J C, DRYER C G, LOWRY C D, et al. Peroxides in gasoline-effects of peroxide formation in cracked gasoline[J].Industrial and Engineering Chemistry, 1934, 26(5): 497-503.
[11] TSEREGOUNIS K C, SCRUGGS B. Deposit formation on a metal surface in oxidized gasoline[J].SOC Autom Enn Trans, Fuels Lubr, 1987, 96 (7): 617.
[12] BATTS B D,FATHONI A Z. A literature review on fuel stability studies with particular emphasis on diesel oil[J].Energy & Fuels, 1991, 5(1): 2-21.
[13] MAYO F R, LAN B Y. Gum and deposit formation from jet turbine and diesel fuels at 100℃[J].Ind Eng Chem Res, 1987, 26(2): 215-220.
[14] PFAENDTNER J, BROADBELT L J. Mechanistic modeling of lubricant degradation:1. Structure-reactivity relationships for free-radical oxidation[J].Ind Eng Chem Res, 2008, 47(9): 2886-2896.
[15] PFAENDTNER J, BROADBELT L J. Mechanistic modeling of lubricant degradation:2. The autoxidation of decane and octane[J].Ind Eng Chem Res, 2008, 47(9): 2897-2904.
[16] MARTIN S M, GRUSE W A, LOWY A. Distribution of gum-forming constituents in cracked gasoline[J].Industrial and Engineering Chemistry, 1933, 25(4): 381-386.
[17] PUENTE G, SEDRAN U. Formation of gum precursors in FCC naphthas[J].Energy & Fuels, 2004, 18(2): 460-464.
[18] PERDEW J P, BURKE K, ERNZERHOF M. Generalized gradient approximation made simple[J].Physical Review Letters, 1996, 77(18): 3865-3868.
[19] PFAENDTNER J, BROADBELT L G. Contra-thermodynamic behavior in intermolecular hydrogen transfer of alkylperoxy radicals[J].Chem Phys Chem, 2007, 8(13): 1969-1978.
[20] PRADELLE F, BRAGA S L, MARTINS F A, et al. Gum formation in gasoline and its blends: A review[J].Energy&Fuels, 2015, 29(12): 7753-7770.
