黔产党参属和金钱豹属植物分子鉴定研究
2018-04-09,
,
(贵阳中医学院药学院, 贵州 贵阳 550025)
党参为我国著名的常用大宗药材,为桔梗科党参属植物党参(Codonopsispilosula)、川党参(C.tangshen)和素花党参(C.pilosulavar.modesta)的干燥根[1]。因党参药食两用和独特的功效越来越受到世人的青睐,近年来已成为用量最多的中药材品种之一。党参属(Codonopsis)植物种类繁多,分类鉴定较为困难。除了药典规定的3个来源,同属多种植物的根也常常混为党参使用,甚至是金钱豹属(Campanumoea)植物的根在贵州等地区也作为“土党参”使用[2]。党参药材常常被加工炮制成饮片或以粉末入药,其性状特征发生了较大变化,采用传统鉴定方法进行准确鉴别较为困难。
自2003年Hebert[3]首次提出DNA条形码(DNA barcoding)概念以来,该技术已成为近年来发展最快,研究最热的一门新兴技术。其通过对所获得的一条或几条DNA片段进行大范围地扫描,构建DNA条形码标准数据库[4],通过比较物种中的一条或少数几条通用的DNA片段就能够对物种进行快速、准确的识别和鉴定,具有简便、快速、可信的巨大优势。Kress和Erickson研究认为,rbcL和psbA-trnH适合植物鉴定[5]。同年,Chase和Cowan等提出,rpoC1、rpoB和matK组合或rpoC1、matK与psbA-trnH组合可用于植物鉴定[6]。Lahaye[7]对兰科植物进行了研究,提出了matK可以鉴定隐藏新物种的观点。2009年Plant Working Group[8]提出matK和rbcL组合鉴定植物。2010年Chen等对6 000余份植物样品进行了DNA条形码筛选,结果表明,ITS 2鉴定效率优于matK和rbcL组合[9]。鉴于党参类药材鉴定较为困难,对党参属和金钱豹属11个物种53份样品的psbA-trnH、rbcL、matK及ITS 2序列进行PCR扩增、测序和Barcoding gap分析,比较各序列的扩增和测序效率,种内和种间变异,并采用BLAST 1和Nearest Distance方法评价不同序列的鉴定能力,以期为该类植物及药材的分类鉴定提供新的参考依据。
1 仪器与材料
1.1 实验仪器
PCR仪(9700,美国ABI公司);低温高速离心机(Centrifuge 5810 R,德国Eppendof公司);凝胶成像系统(GGM/D 2,英国SYNGENE);电泳仪(DYY-6 C,北京六一仪器厂);自动蒸汽灭菌锅(ES-315,深圳市博大精科技实业有限公司);微型漩祸混合仪(WH-1,常州易晨仪器制造有限公司)。
1.2 实验试剂
新型植物基因组DNA提取试剂盒(DP-320离心柱型,Tiangen公司);引物(上海生工生物工程有限公司);2×TapPCR MasterMix(KT 201,Tiangen公司);Marker(D 2000,Tiangen公司);5×TBE电泳缓冲液(SD 8102,上海生工生物工程有限公司);Goldview I型核酸染色试剂(D 0125,Solarbio);琼脂糖(SunShineBio,南京生兴生物技术有限公司)。
1.3 实验样品
本研究所有样品均采集于贵州省、云南省、四川省,共53份,经贵阳中医学院孙庆文教授鉴定,凭证标本存放于贵阳中医学院生药实验室,实验材料见表1。
表1样品来源
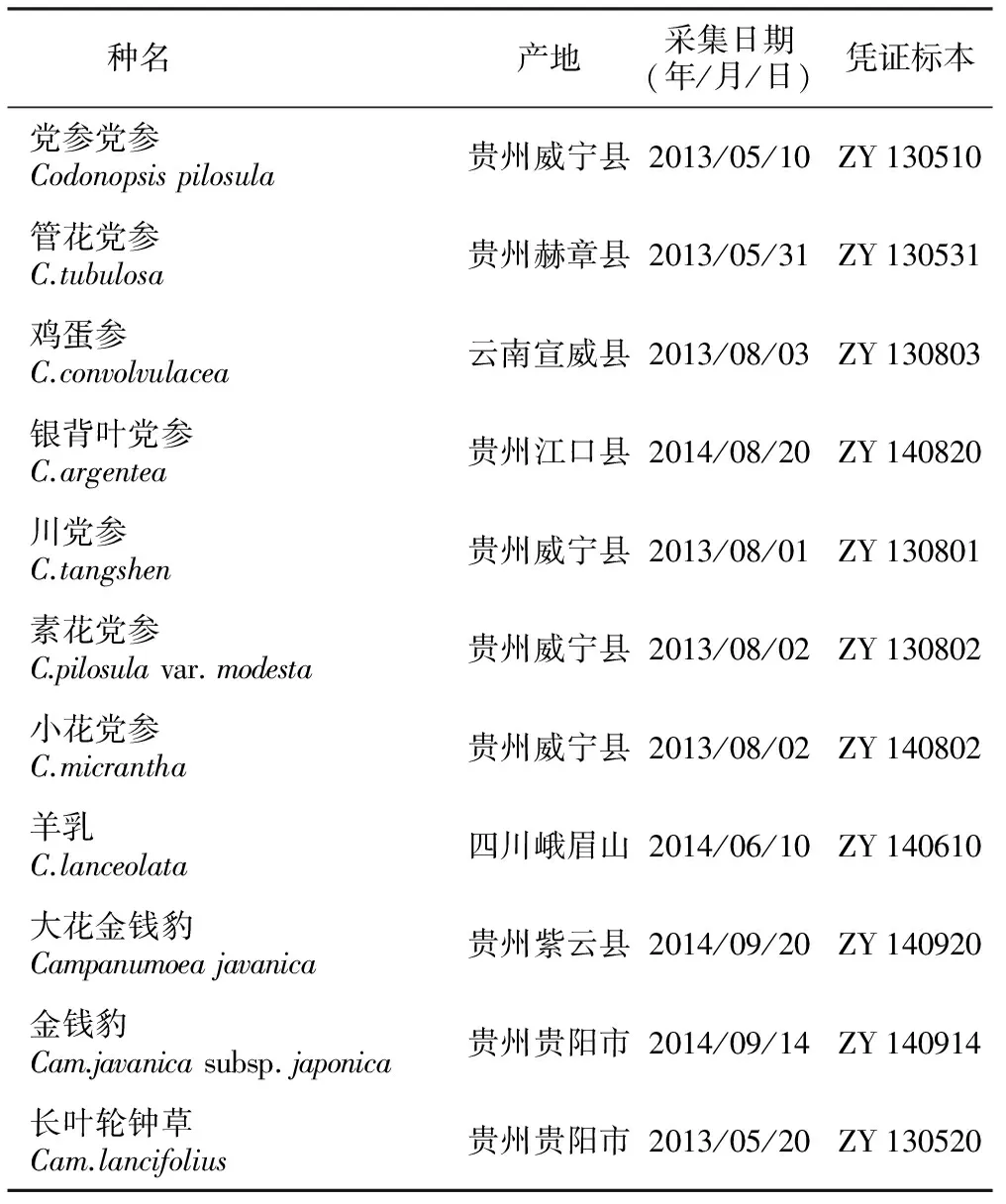
种名产地采集日期(年/月/日)凭证标本党参党参Codonopsispilosula贵州威宁县2013/05/10ZY130510管花党参C.tubulosa贵州赫章县2013/05/31ZY130531鸡蛋参C.convolvulacea云南宣威县2013/08/03ZY130803银背叶党参C.argentea贵州江口县2014/08/20ZY140820川党参C.tangshen贵州威宁县2013/08/01ZY130801素花党参C.pilosulavar.modesta贵州威宁县2013/08/02ZY130802小花党参C.micrantha贵州威宁县2013/08/02ZY140802羊乳C.lanceolata四川峨眉山2014/06/10ZY140610大花金钱豹Campanumoeajavanica贵州紫云县2014/09/20ZY140920金钱豹Cam.javanicasubsp.japonica贵州贵阳市2014/09/14ZY140914长叶轮钟草Cam.lancifolius贵州贵阳市2013/05/20ZY130520
2 方法与结果
2.1 实验方法
2.1.1总DNA提取、凝胶电泳、PCR扩增及测序
选取硅胶干燥的植物叶片约20 mg,采用试剂盒提取总DNA。PCR反应体系50μL,体系包含2μL总DNA(约30 ng)、26μL 2×TapPCR MasterMix、引物2μL(2.5μmol/L)和20μL ddH2O。各个标记的扩增引物及扩增程序见表2。取5μL PCR扩增产物,以Marker作为标记于1.2%的凝胶板上电泳30 min检测,于凝胶图像分析系统下观察,拍照(见图1),将获得的单一明亮条带产物送生工生物工程有限公司进行测序。
2.1.2数据处理
测序峰图采用序列拼接软件CodonCode AlignerV 5.12(CodonCode Co.,USA)校对拼接,去掉引物区及低质量区,所得各序列经MEGA 5.0软件进行多序列比对、查错;采用 PCR 扩增效率和测定成功率评价各个分子标记的扩增及测序情况;利用Song等[10]的方法,计算遗传距离K 2 P值,比较不同标记基因在种间、种内变异幅度,最后采用Ross等[11]的比对法和最小距离法考察标记基因的鉴定效率。
表2各引物名称、扩增程序及反应体系

标记基因引物名引物序列5′⁃3′扩增条件反应体系ITS22FGCGATACTTGGTGTGAAT3RGACGCTTCTCCAGACTACAAT94℃5min,94℃30s,56℃30s,72℃45s,40cycles,72℃10minMix13μL,引物1μL,总DNA1μL,ddH2O9μLpsbA⁃trnHfwdGTTATGCATGAACGTAATGCTCrevCGCGCATGGTGGATTCACAATCC94℃5min,94℃1min,55℃1min,72℃45s,30cycles,72℃1minMix13μL,引物1μL,总DNA1μL,ddH2O9μLmatK3F_KIMCGTACAGTACTTTTGTGTTTACGAG1R_KIMACCCAGTCCATCTGGAAATCTTGGTTC95℃4min,94℃30s,56℃45s,72℃45s,35cycles,72℃10minMix13μL,引物1μL,总DNA3μL,ddH2O7μLrbcLLACGTAGCTTATCCTTTAGACCHTGGCATAGAGACCCAATCTT94℃5min,94℃1min,55℃1min,72℃45s,30cycles,72℃1minMix13μL,引物1μL,总DNA3μL,ddH2O7μL

图1 DNA序列部分电泳图
2.2 结 果
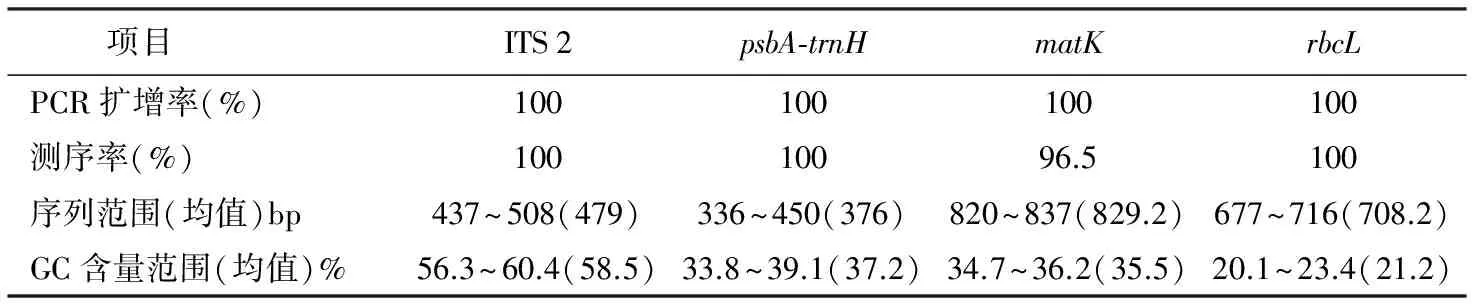
2.2.1PCR扩增及测序率、序列长度及GC含量
PCR扩增率和测序率是评价DNA条形码序列的一个重要指标。各序列扩增率均为100%。测序率除了matK为96.5%外,其余测序率为100%。序列长度考察发现,matK最长,其余依次是rbcL、ITS 2和psbA-trnH。同时ITS 2和psbA-trnH序列长度在种间和种内差异较大。rbcL在种间和种内有一定变化,matK序列长度几乎无变化,详见表3。
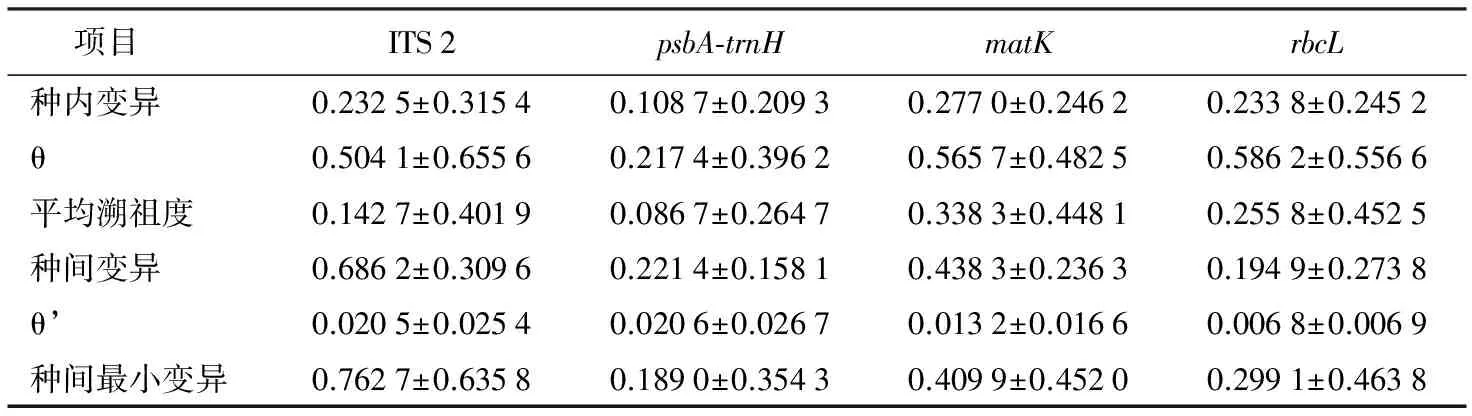
2.2.2种内及种间变异分析
最理想的条形码是种内最大遗传变异应小于种间最小遗传变异,并且两者之间有显著差异。分析序列种间和种内变异情况,结果表明,ITS 2种间变异最大,其余依次是matK、psbA-trnH和rbcL,且ITS 2和matK明显大于psbA-trnH和rbcL。种内变异不大,依次是matK、rbcL、ITS 2和psbA-trnH,详见表4。
2.2.3不同序列Barcoding gap分析
一条理想的DNA条形码是种间差异足够大,种内差异尽量小,并且在两者间形成一个间隔区。从Barcoding gap种间、种内的分布图来看,4条DNA条序列的Barcoding gap均不明显,种间、种内变异分布呈一边分散趋势,不利于该类植物鉴定,见图2。
表3DNA序列长度及GC含量
项目ITS2psbA⁃trnHmatKrbcLPCR扩增率(%)100100100100测序率(%)10010096.5100序列范围(均值)bp437~508(479)336~450(376)820~837(829.2)677~716(708.2)GC含量范围(均值)%56.3~60.4(58.5)33.8~39.1(37.2)34.7~36.2(35.5)20.1~23.4(21.2)
表44个序列种间种内差异
项目ITS2psbA⁃trnHmatKrbcL种内变异0.2325±0.31540.1087±0.20930.2770±0.24620.2338±0.2452θ0.5041±0.65560.2174±0.39620.5657±0.48250.5862±0.5566平均溯祖度0.1427±0.40190.0867±0.26470.3383±0.44810.2558±0.4525种间变异0.6862±0.30960.2214±0.15810.4383±0.23630.1949±0.2738θ’0.0205±0.02540.0206±0.02670.0132±0.01660.0068±0.0069种间最小变异0.7627±0.63580.1890±0.35430.4099±0.45200.2991±0.4638
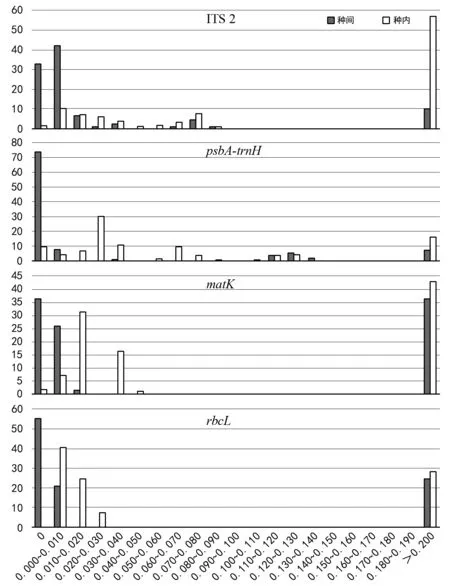
图2 DNA条序列的Barcoding gap
2.2.4候选序列鉴定率评价
采用对比法和最小距离法评价4条序列的鉴定率,结果表明,rbcL鉴定率最高,其余依次是ITS 2、matK和psbA-trnH。详见表5。
3 讨 论
3.1 序列筛选
DNA条形码是利用基因组中一条公认的、标准的、相对较短的DNA片段来进行物种鉴定的分子鉴定技术。线粒体基因组进化最慢,对植物分辨率较低,适合动物分子鉴定,不适合植物分子鉴定。相对于线粒体基因,叶绿体基因进化相对较快,适合植物分子鉴定。核基因组进化最快,也适合植物分子鉴定,所以叶绿体基因组和核基因组常用于植物分子鉴定。但叶绿体基因组和核基因组不同序列对各个植物类群又有差异。因此寻找适合各科属植物鉴定的DNA条形码成为现阶段研究的焦点。本实验选择目前认为在各个科属通用性较好的条形码来考察党参属及其近缘属的分子鉴定方法。ITS 2为核基因组,是rDNA上位于5.8 S和26 S之间的一段进化速度很快的非编码区,物种间变异位点较多,长度很短,具有较高的扩增效率和物种鉴定效率。matK、rbcL和psbA-trnH位于叶绿体基因组,由于其进化速率较快,并且有较多的变异位点,常用于植物的分子鉴定,但3条序列侧重点不同,rbcL适合分类阶元较高的类类群,matK和psbA-trnH则恰好相反。考虑到实验材料涉及到不同属,所以选择了3条叶绿体基因作为党参属金钱豹的分子鉴定。
3.2 PCR扩增及测序率
PCR扩增及测序率是进行后续分类鉴定的前提。本实验中,4条序列的PCR扩增及测序率较高,基本达100%,说明所选序列在党参属和金钱豹属植物中扩增及测序较好,这为后面的序列分析提供了可能。
表5对比法和最小距离法分析4条候选序列的鉴定效率

序列鉴定方法样品数鉴定成功率rbcL对比法4397.9最小距离法43100.0matK对比法5389.7最小距离法5387.9ITS2对比法5182.1最小距离法51100.0psbA⁃trnH对比法5382.8最小距离法5360.3
3.3 种间、种内变异及Barcoding gap评估
一条好的DNA条形码应具有足够高的种间多样性,用于区分不同物种。种间变异、θ’及最小种间距离用于测定种间差异[12-13]。分析结果表明,ITS 2种间变异最大,显示出其种间最高的多样性,其次是matK,最小的是rbcL。种内变异、θ及最大种内距离用于种内差异,matK种内变异最大,psbA-trnH种内变异最小。
理想的DNA条形码是遗传变异种间和种内变异明显分开,不存在重叠现象。根据Barcoding gap图,4条DNA序列重叠率均较大,不能将党参属及金钱豹属植物分开。Meyer等[12]和Moritz等[14]用实验证实了密切相关的种增加时种内和种间的遗传变异交叉重叠现象会增加,本实验可能是由于党参属与金钱豹属植物比较近缘,并且每个种数量较多,因此导致其无明显的gap区。
3.4 鉴定率评估
DNA条形码物种鉴定方法目前主要有对比法(BLAST 1)、最小距离法(Nearest distance)和建树法(Tree-based method)。本实验采用对比法、最小距离法用于评价各条序列的鉴定成功率,主要是为了更好地考察各条序列的鉴定能力。对比法[15]是基于相似度的方法将鉴定DNA序列与数据库DNA序列进行两两局部比对或搜索短的核苷酸字符串来查找数据库中与之最相似的DNA序列。4条DNA序列对比法鉴定结果显示,rbcL鉴定率最高,其次是matK,最低的是psbA-trnH。最小距离法[15]是将鉴定DNA序列与参考DNA序列进行两两比对,当参考DNA序列与鉴定DNA序列有最小的两两比对距离时,则可进行距离判定。4条序列最小距离法鉴定显示,rbcL和ITS 2鉴定率最高,达到100%,psbA-trnH最低,仅60.3%。使用2种鉴定方法来评价4条序列的鉴定能力,结果发现,rbcL和ITS 2使用最小距离法鉴定效果较好,rbcL和matK使用对比法较佳。综上所述,rbcL和ITS 2可用于党参属及其近缘属的分子鉴定,matK可作为候选条形码序列。
参考文献:
[1]国家药典委员会.中国药典(2015年版一部)[S].北京:中国医药科技社,2015:281-282.
[2]孙庆文,黄敏,何顺志.高效液相色谱法测定土党参中党参炔苷的含量[J].时珍国医国药,2009,20(1):120-121.
[3]Hebert P D N,Ratnasingham S,deWaard J R.Barcoding animal life:cytochrome coxidase subunit 1 divergences among closely related species[J].Proc R Soc Lond B Biol Sci,2003,270:96-99.
[4]Kress WJ,Erickson DL.DNA barcodes:Genes,genomics,and bioinformatics[J].Proceedings of the NationalAcademy NationalAcademy of Sciences,USA,2008,105:2 761-2 762.
[5]Kress W J,Erickson D L.A two-locus global DNA barcoding for land plants:the codingrbcLgene complements the non-codingtrnH-psbAspacer region[J].PLOS ONE,2007,2:e 508.
[6]Chase M W,Cowan R S,Hollingsworth P M,et al.A proposal for a standardised protocol to barcode all land plants[J].Taxon,2007,56:295-299.
[7]Lahaye R,van der Bank M,Bogarin D,et al.DNA barcoding the floras of biodiversity hot spots[J].Proc Natl Acad Sci USA,2008,105:2 923-2 928.
[8]CBOL Plant Working Group.A DNA barcoding for land plants[J].Proc Natl Acad Sci USA,2009,106:12 794-12 797.
[9]Chen S L,Yao H,Han J P,et al.Validation of the ITS 2 region as a novel DNA barcoding for identifying medicinal plant species[J].PLOS ONE,2010,5:e 8 613.
[10]Song J Y,Yao H,Li Y,et al.Authentication of the family Polygonaceae in Chinese Pharmaeopoeia by DNA barcoding technique[J].Journal of Ethnopharmacol,2009,124(3):434-439.
[11]Ross H A,Murugan S,Li W L.Testing the reliability of genetic methods of species ideniification via simulation[J].Syst Biol,2008,57:216-230.
[12]Meyer C P,Paulay G.DNA barcoding: error rates based on comprehensive sampling[J].PLOS Biol,2005,3:e 422.
[13]Meier R,Zhang G Y,Ali F.The use of mean instead of smallest interspecific distances Exaggerates the Size of the“Barcoding Gap”and Leads to Misidentification[J].Syst.Biol.2008,57(5):809-813.
[14]Moritz C,Cicero C.DNA barcoding: promise and pitfalls[J].PLoS Biol,2004,2:e 354.
[15]陈士林.中药DNA条形码分子鉴定[M].北京:人民卫生出版社,2012:19-20.
