新疆葡萄和葡萄酒相关酿酒酵母种群的结构特征
2018-04-08贾佳,王冠群,朱丽霞,韩培杰,郭东起,陈明
贾 佳, 王 冠 群, 朱 丽 霞, 韩 培 杰, 郭 东 起, 陈 明
( 1.大连工业大学 生物工程学院, 辽宁 大连 116034;2.塔里木大学 生命科学学院, 新疆 阿拉尔 843300;3.中国科学院微生物研究所 真菌学国家重点实验室, 北京 100101 )
0 引 言
酿酒酵母最早的应用起源于葡萄酒的酿造,伴随人类文明发展而成为经过几千年驯化的一种微生物[1]。随着现代生物技术的不断更新,例如inter-delta序列扩增、微卫星技术、多位基因测序乃至全基因组分,来自世界各地不同环境的酿酒酵母不断地被科学证实其多样性十分丰富,且种群内地域分化特征十分明显,与人类的活动密切相关[2]。随着酿酒酵母与葡萄种植和葡萄酒酿造等生态环境相关性研究的深入,受葡萄品种、葡萄生长地区的气候条件及葡萄种植等人类活动的影响,酿酒酵母种间遗传特性被证明具有显著差异[3-4],通过其种群之间进化关系,推测其可能起源于地中海地区,人类活动是其重要的传播途径[5]。
葡萄作为新疆主要作物之一,品种丰富且种植历史悠长,但在现代农业经济利益的驱动下,长期种植的本地葡萄品种种类与种植面积急剧减少,而新引进品种的快速与面积的急剧扩,严重影响了地方传统农业种植生态环境。本实验通过delta基因片段和微卫星分析对新疆葡萄及葡萄酒相关的酿酒酵母的种内多态性和地域类群分化进行研究,推测其种群结构形成的可能原因。
1 实 验
1.1 实验菌株
挑选覆盖35地区的296株与葡萄和葡萄酒相关酿酒酵母(下文简称新疆酿酒酵母)用于delta分析酵母种群的多样性和区域相关性,其中来自29个地区的220株菌株用于微卫星分析酿酒酵母菌株的区域相关性。
1.2 DNA提取及PCR扩增
将新鲜待测菌株进行DNA提取,提取方法参考文献[6]。delta基因片段扩增引物为delta12(5′-TCAACAATGGAATCCCAAC-3′)和delta21(5′-CATCTTAACACCGTATATGA-3′),退火温度改为44 ℃,其余条件同文献[7]。SSR分析选用2组12个位点(C3、C5、C8、C9、C11、SCYOR267C和YKL172W、SCAAT1、C4、SCAAT5、C6、YPL009C)的组合进行多重PCR扩增,反应体系及扩增条件见参考文献[1]。
1.3 数据分析
基因delta区域PCR产物的凝胶电泳条带用凝胶专业分析软件(4.0.00.001版)统计并转化形成0-1数据矩阵。SSR的PCR产物通过毛细管电泳验证,并将图谱条带归一化并转换成0-1数据;用软件POPGENE 1.32在物种和变种水平上分别估测下列遗传参数:多态位点百分比(PPL)、Shannon信息指数(Is)、观测等位基因数(Ao)、有效等位基因数(Ae)、Nei’s基因多样性(He)、总基因多样系数(Ht)、居群内多样性系数(Hs)、居群间分化系数(Gst),居群间基因漂流值基因流(Nm)、Nei’s 无偏见遗传一致性和遗传距离,将相应的数据矩阵导入NTSYS-pc 2.10统计软件,根据Nei的方法计算遗传相似系数,按不加权成对算术评价法(UPGMA)进行遗传相似性聚类;对剔除相同带型菌株后的0-1矩阵进行分别用Phyltools(1.32版)和Phylip(3.695版)构建菌株水平的进化树,再将进化树中每个分支出现的地区标注为1,未出现的记为0,构建酿酒酵母分支和地区二元矩阵利用多维标度分析的(MDS)关联图(Orange canvas 2.70),压力函数选用Signed relative stress。
2 结果与分析
2.1 酿酒酵母多样性分析
如表1所示,220株酿酒酵母中,有192个不同基因型,其多态性明显高于delta基因分型(296株中有125个基因型),南疆和北疆delta基因型个数占总菌数比例接近一致,分别为0.690和0.698,所得的基因型和总待测菌数之比也接近相等0.88和0.86,说明SSR基因分型效果明显高于delta基因分型。对SSR基因及delta进行PPL、Is、Ao、Ae、He、Hs、Ht、Gst分析,得到两种基因型表现出新疆葡萄与葡萄酒酿酒酵母在地域群体内部还是群体之间具有较高的遗传多样性,群居间基因流,delta表现出更丰富的基因多态性,SSR反映出酿酒酵母居群之间具有较高的分化,自然在居群间基因流偏小。
2.2 新疆酿酒酵母地域群体分化特征
基于已获得的酿酒酵母delta和SSR基因型,构建酿酒酵母的地域群体进化树,如图1、2所示。基于delta基因型和SSR基因型的两种群体进化树能将北疆和南疆多数地区聚在一起。从图中可以看出,南北疆界限明显,各地域之间分化较明显,几乎一个地域一个分支。从树根部到树顶部,南疆各地域之间具有明显的进化层次,但北疆地区具有明显的地区聚集效应,两种基因型的地域类群进化树中每个类群或亚类群中的地域构成不同。同时在对所有菌株进行PCA聚类分析,获得同样的结果,南北疆类群具有明显分化特征。

表1 基于SSR和inter-delta的新疆葡萄与葡萄酒酿酒酵母基因多态性分析Tab.1 The genetic diversity of S. cerevisiae associated with grape and wine in Xinjiang based on SSR and inter-delta

图1 新疆29个地区的酿酒酵母基于SSR基因型的类群结构图
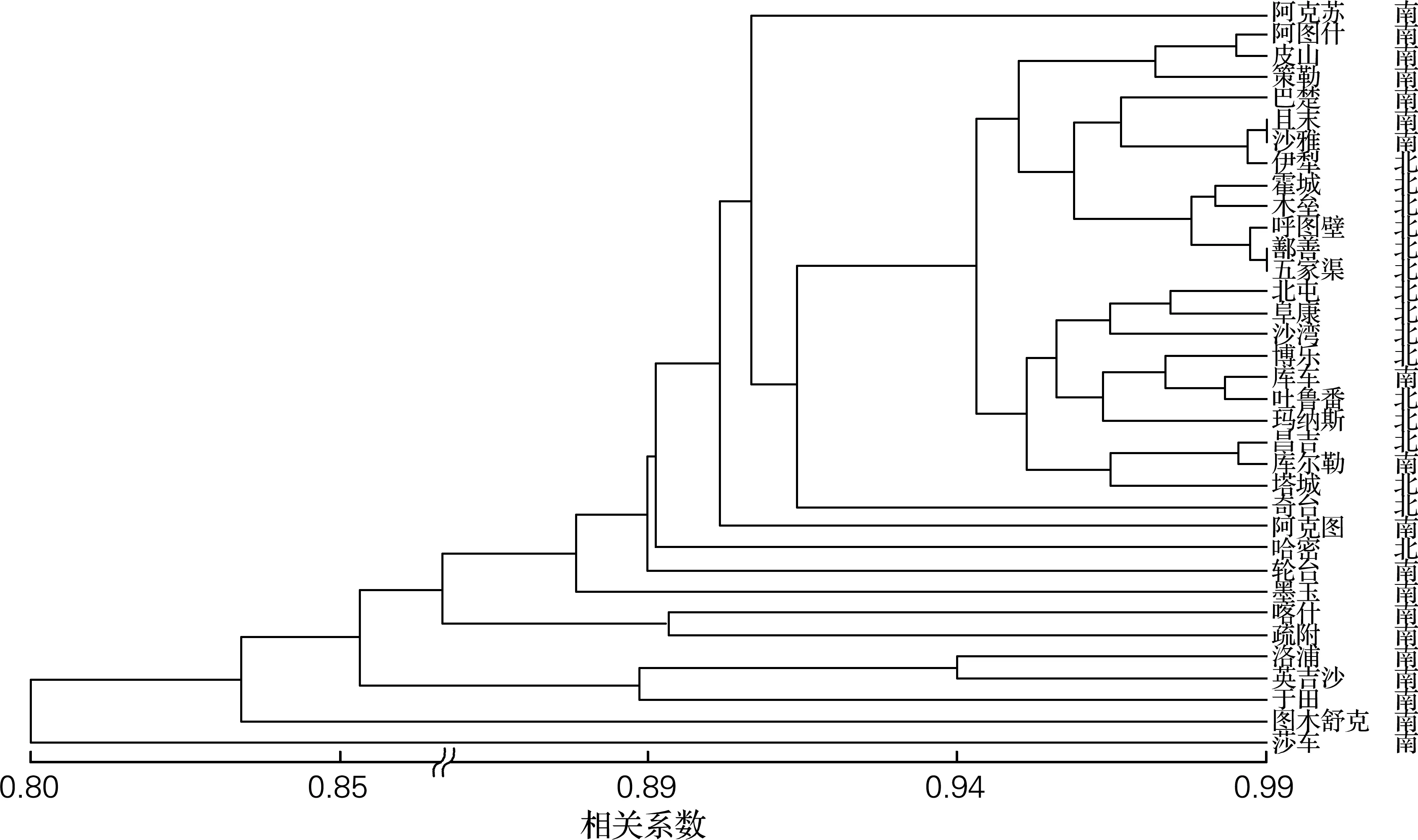
图2 新疆35个地区的酿酒酵母基于delta基因型的类群结构图
2.3 新疆酿酒酵母地区群落之间的联系及其扩散途径
基于delta的125个基因型和SSR的192个基因型,分别构建新疆葡萄和葡萄酒相关的酿酒酵母的进化树,依据进化树中每个分支中菌株来源地,将其归类为南疆分支群(SX)、北疆分支群(NX)及南北疆分支混合群(mNSX)。delta进化树中共有55个分支构成,而SSR进化树中38个分支构成,分别按照“1.3”的方法构建分支和地区二元矩阵,由Orange canvas 2.7软件进行多维标度分析(MDS),结果如图3、4所示。从图中可以看出,模型与预期地域类群具有高拟合度(模型压力分别为0.263和0.256),绝大多数南疆地域类群具有较强的关联性,其中心位置为墨玉、疏附、洛浦、阿图什、阿克陶、于田、英吉沙、沙雅、图木舒克等。将南疆与北疆联系起来的有哈密、吐鲁番、阜康、伊犁、库车、轮台、库尔勒地区,其中哈密地区与南疆具有比较紧密的联系。

图3新疆35个地区的酿酒酵母基于delta基因型的地域类群关联图(圆点大小与透明度表示压力大小,红色为南疆地区,蓝色为北疆地区)
Fig.3The geographical relations of 35 kinds ofS.cerevisiaeregional populations in Xinjiang based on MDS from the data of inter-delta genotypes (the stress value of different plots were shown by different size and transparency)
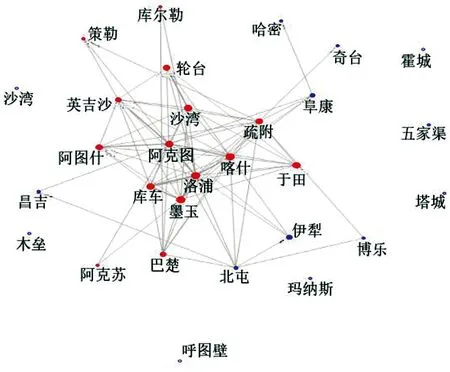
图4新疆29个地区的酿酒酵母基于SSR基因型的地域类群关联图(圆点大小与透明度表示压力大小,红色为南疆地区,蓝色为北疆地区)
Fig.4The geographical relations of 29 kinds ofS.cerevisiaeregional populations in Xinjiang based on MDS from the data of inter-delta genotypes (the stress value of different plots were shown by different size and transparency)
北疆的各地区之间的联系十分松弛,将北疆各地方类群单独进行MDS分析,如图5、6所示。

图5北疆15个地区酿酒酵母基于inter-delta基因型的地域类群关联图(圆点大小与透明度表示压力大小)
Fig.5The geographical relations of 15 kinds ofS.cerevisiaeregional populations in northern Xinjiang based on MDS from the data of inter- delta genotypes (the stress value of different plots were shown by different size and transparency)

图6北疆18个地区酿酒酵母基于inter-delta基因型的地域类群关联图(圆点大小与透明度表示压力大小)
Fig.6The geographical relations of 18 kinds ofS.cerevisiaeregional populations in northern Xinjiang based on MDS from the data of inter-delta genotypes (the stress value of different plots were shown by different size and transparency)
以与哈密、吐鲁番、奇台、阜康、昌吉、伊犁、北屯为中心,向其他地区辐射,但依然不能将所有地域联系起来,且地理位置相近的有些地区并没有相关的联系。
3 讨 论
3.1 Inter-delta和SSR基因分型的可行性
Inter-delta和SSR对S.cerevisiae在菌株水平上具有很好的区分效果[8-9],本实验中的delta基因能将近70%的酿酒酵母菌株进行有效分型,SSR能将近90%的菌株进行有效分型,且分型明显效果优于delta,其原因之一为SSR所选用的引物对数远远高于delta,对比的基因片段累计长度远远大于inter-delta基因片段长度,能够捕捉更多的基因突变点。在最初样本进行分离时,南疆样品中的疑似酿酒酵母相当少,有些地方没有分离到,而北疆地区相对多,由此限制了本实验中南北疆所测试的酿酒酵母菌株数不一致,但利用两种基因分型技术,得到的南北疆有效分型菌数占总菌数的比例接近一致,表明所选酿酒酵母的样本数量满足本实验的相关研究。
3.2 新疆酿酒酵母与地域特征的关联性
目前酿酒酵母多样性形成、种群的分化及分布归因于地理位置、环境及人类活动共同的相互作用[10-12]。在新疆,三山夹两盆的地貌特征使得新疆形成南、北、东疆截然不同的地貌与气候特征。在葡萄种植方面,南疆地区为农家小院式种植,主要品种为种植历史比较悠久的木纳格、和田红、马奶子等地方葡萄品种。而北疆地区为大面积种植方式,沿天山北带地区(昌吉、玛纳斯、阜康、沙湾等)、伊犁地区多数为酿酒葡萄,霍城、塔城地区、北屯、博乐等地区为单一红提鲜食葡萄,哈密、吐鲁番地区为地方无核白。这些复杂而差异巨大的气候条件及人类等活动,导致新疆酿酒酵母菌种内基因多态性和地域种群有限分化,种群结构南北疆界限明显,南疆内部联系紧密,且具有较清晰的进化层次,主要原因可能是天山的物理阻隔和葡萄的种植方式,南疆西部的喀什和和田部地域在delta和SSR基因进化上表现较为久远,这极大可能缘于喀什、洛浦、墨玉、阿克陶、阿图什、疏附等这些地域,被塔克拉玛干沙漠、昆仑山及天山所包围,形成一个相对独立的生态空间,如图7所示。

图7新疆酿酒酵母种群扩散图(线的粗细表示联系的紧密程度)
Fig.7The geographical relationships ofS.cerevisiaepopulation from Xinjiang (the width of line means relationship tightness between regions)
酿酒酵母在北疆各地区之间没有明显的进化层次很有可能与引进葡萄品种的种植历史较短及大面积现代化种植方式有关,进一步证明了新疆酿酒酵母的菌群结构特征明显受到人类活动和地域气候等的多重作用[5]。
3.3 新疆酿酒酵母种群扩散模式分析
利用有限数据构建的地域类群进化树及多维标度法构建的关联图,绘制形成酿酒酵母可能的传播途径示意图,如图7所示。可以看到,新疆酿酒酵母从南疆的喀什、和田地区向北经过沙雅、库车、轮台、库尔勒扩散至北疆的哈密,以此向西扩散至吐鲁番、阜康、奇台,再由奇台向北连接北屯,向西连接阜康、昌吉、伊犁及等沿途地区。这种扩散模式与新疆的三条丝绸之路中北路和中路具有惊人的相似。因此,新疆酿酒酵母的群体结构特征的形成,可能与人类历史上的丝路文明传播有关。这虽然与Knight[13]在研究中基于SSR分析得到新西兰葡萄与葡萄酒相关酿酒酵母种群地域之间的联系与分化程度与新西兰葡萄酒工业的影响有类似性,但基于数据和方法的有限性,同时欠缺年份等其他因素的影响,对此结论需要更大系统性的样本量和先进的技术,例如基因组进行深入论证与修正。
4 结 论
基因delta对酿酒酵母的分型率能达到70%,而SSR分型率达到90%,delta能够较好地揭示酿酒酵母在地域类群内部及整体基因水平的遗传多样性,而SSR在地域类群之间分化水平的评估比delta效果好。新疆酿酒酵母种内多态性十分丰富和地域居群之间分化明显,几乎每个地区居群都成为一个相对独立的分支,南北疆大菌群之间分化十分明显,南疆内部联系紧密,且具有从西到东,从远到近的较清晰的进化层次;而北疆地区联系十分疏松,北疆内部各地区没有明显的进化层次。酿酒酵母的菌群结构特征明显受到地方多变的气候特征、明显不同的葡萄品种、种植方式等多重作用的影响,还可能与丝绸之路上的人类文明传播活动有关。
参考文献:
[1] LEGRAS J L, MERDINOGLU D, CORNUET J M, et al. Bread, beer and wine:Saccharomycescerevisiaediversity reflects human history[J]. Molecular Ecology, 2007, 16(10): 2091-2102.
[2] SCHACHERER J, SHAPIRO J A, RUDERFER D M, et al. Comprehensive polymorphism survey elucidates population structure ofSaccharomycescerevisiae[J]. Nature, 2009, 458(7236): 342-345.
[3] AYOUB M J, LEGRAS J L, SALIBA R, et al. Application of multi locus sequence typing to the analysis of the biodiversity of indigenousSaccharomycescerevisiaewine yeasts from Lebanon[J]. Journal of Applied Microbiology, 2006, 100(4): 699-711.
[4] SIPICZKI M. Diversity, variability and fast adaptive evolution of the wine yeast (Saccharomycescerevisiae) genome—a review[J]. Annals of Microbiology, 2010, 61(1): 85-93.
[5] KNIGHT S, KLAERE S, FEDRIZZI B, et al. Regional microbial signatures positively correlate with differential wine phenotypes: evidence for a microbial aspect toterroir[J]. Nature Scientific Report, 2015, 5: 14233-14259.
[6] AMBERG D C, BURKE D, STRATHERN J. Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual[M]. New York: Cold Spring Harbor Laboratory Press, 2005.
[7] LEGRAS J L, KARST F. Optimisation of interdelta analysis forSaccharomycescerevisiaestrain characterisation[J]. FEMS Microbiology Letters, 2003, 221(2): 249-255.
[8] EZERONYE O U, LEGRAS J L. Genetic analysis ofSaccharomycescerevisiaestrains isolated from palm wine in eastern Nigeria. Comparison with other African strains[J]. Journal of Applied Microbiology, 2009, 106(5): 1569-1578.
[9] PATHANIA N, KANWAR S S, JHANG T, et al. Application of different molecular techniques for deciphering genetic diversity among yeast isolates of traditional fermented food products of western Himalayas[J]. World Journal of Microbiology and Biotechnology, 2010, 26(9): 1539-1547.
[10] WANG Q M, LIU W Q, LITI G, et al. Surprisingly diverged populations ofSaccharomycescerevisiaein natural environments remote from human activity[J]. Molecular Ecology, 2012, 21(22): 5404-5417.
[11] EBERLEIN C, LEDUCQ J B, LANDRY C R. The genomics of wild yeast populations sheds light on the domestication of man’s best (micro) friend[J]. Molecular Ecology, 2015, 24(21): 5309-5311.
[12] HYMA K E, FAY J C. Mixing of vineyard and oak-tree ecotypes ofSaccharomycescerevisiaein North American vineyards[J]. Molecular Ecology, 2013, 22(11): 2917-2930.
[13] KNIGHT S, GODDARD M R. Quantifying separation and similarity in aSaccharomycescerevisiaemetapopulation[J]. The ISME Journal, 2015, 9: 361-370.
