Carbon deposition and catalytic deactivation during CO2 reforming of CH4 over Co/MgO catalyst☆
2018-04-08JianweiLiJunLiQingshanZhu
Jianwei Li ,Jun Li ,Qingshan Zhu
1 State Key Laboratory of Multiphase Complex Systems,Institute of Process Engineering,Chinese Academy of Sciences,Beijing 100190,China
2 State Key Laboratory of Heavy Oil Processing,China University of Petroleum,Beijing 102249,China
3 School of Chemical Engineering,University of Chinese Academy of Sciences,Beijing 100049,China
Keywords:Co/MgO CH4 reforming Carbon deposition Co oxidization
A B S T R A C T The deactivation mechanism of Co/MgO catalyst for the reforming of methane with carbon dioxide was investigated.The conversion of CH4 displayed a significant decrease in the initial stage caused by carbon deposition.There were two types of cokes,carbon nanotubes(CNTs)and carbon nano-onions(CNOs).The number of the CNO layers that coated on the surface of Co nanoparticles(NPs)increased rapidly in the initial reforming time,which was responsible for the deactivation of the Co/MgO catalyst.The deposition of CNOs was attributed to the oxidation of Co NPs.Therefore,the deactivation of the Co/MgO catalyst was originated from the first oxidization of the Co NPs into Co3O4 by O species(OH intermediate,CO2,H2O)during the reforming reaction,which accelerates the formation of coke that blocked the active metal,thus led to catalyst deactivation.
1.Introduction
CO2reforming of methane has attracted much attention in recent years,because this process is able to convert the two greenhouse gases of CH4and CO2to syngas with low H2/CO ratios which could be directly used for the Fischer–Tropsch process[1–4].Various supported metal catalysts have been developed including noble metal and transition metal catalysts.Generally,noble metals like Pt[5–7],Ru[8–10]and Pd[11,12]have been reported to have high activity and stability during the CO2–CH4reforming reaction.However,their commercial use is limited by the high cost of noble metals.Compared to the noble-metal catalysts,Co-based catalysts are more suitable for this process due to the relatively high activity and lower cost.However,the main problem of Co-based catalysts to be applied to industry is the rapid deactivation originated from severe carbon deposition and sintering of metal nanoparticles(NPs)under high reaction temperatures[13–16].Therefore,promoting Co-based catalysts on various supports like ZrO2[17,18],TiO2[19,20],and MgO[21,22]has been introduced to enhance anti-sintering and coke resistance performance of Co-based catalysts.MgO has been proved to be an effective support for stabilizing the Co NPs due to the formation of solid solution(CoMgOx)[21,22]and providing strong Lewis basicity[23,24],which has a strong adsorption for CO2to resist carbon deposition.Particularly,the exposed(111)surface of MgO creates a strong electrostatic field perpendicular to itself due to the structure of alternating polar nanoplayers of O2-and Mg2+and there are a large amount of medium O2-Lewis basic sites on MgO(111)surface.Previous research has reported that the deactivation of Co NPs supported on Al2O3[17,25,26]and SiO2[27,28]catalysts is caused by carbon deposition and sintering of Co NPs.In addition,Ruckenstein and Wang[29]also find that Co/Al2O3with low metal loading can be oxidized during the CH4–CO2reforming process.However,the relationship between carbon deposition behavior and metal oxidization has not been reported yet.
In this contribution,the relationship between carbon deposition behavior and metal oxidization was investigated over a Co/MgO catalyst for production of syngas via CH4–CO2reforming.The results showed that the Co/MgO catalyst exhibited strong anti-sintering performance and metal sintering was not responsible for catalyst deactivation.Meanwhile the deactivation and significant carbon deposition were attributed to oxidization of Co NPs which accelerated CNO formation totally coating active metal and leads to deactivation.
2.Experimental
2.1.Catalyst preparation
The 10 wt%(metal loading)Co/MgO catalyst was prepared by impregnation method using raw materials of Co(NO3)2·6H2O(AR grade,J&K.Corp)and MgO(AR grade,J&K.Corp).Generally,Co(NO3)2·6H2O was solved in deionized water to form a Co(NO3)2solution.MgO(pretreated at 500°C for 3 h before using)was impregnated with the Co(NO3)2solution,followed by drying at 100°C for 12 h and calcination at 500°C for 5 h.The obtained CoO/MgO catalyst was ground and sieved to select the particle size of 70–150 μm with a bulk density of 600 kg·m-3.
2.2.Catalyst characterization
The phase structure of the catalysts was analyzed using X-ray diffractometry(XRD,X'Pert MPD Pro,Panalytical,Netherlands)with Cu Kαradiation(λ=0.15408 nm)at 40 kV and 40 mA.The morphology of the catalysts was characterized using a JEM-2100 transmission electron microscope(TEM)operated at 200 kV.
Temperature programmed reduction of hydrogen(H2-TPR)was performed on a chemisorption apparatus(ChemBet 3000,Quantachrome,USA).The sample(30 mg)was pretreated with Ar(99.99%)flow at 200°C for 30 min,followed by cooling it to room temperature.Then the sample was heated to 900°C at a constant heating rate of 10°C·min-1using a flow of H2/Ar mixture(volume ratio,10:90)under a flow rate of 30 ml·min-1.The signal of hydrogen consumption was detected by a thermal conduction detector(TCD).
The amount of deposited carbon on the spent catalysts was measured by thermogravimetry(TG,STA 449C,Netzsch,Germany).Before the test,the sample(about 30 mg)was pretreated by N2(99.99%)at 300°C for 30 min,cooled down to room temperature,followed by a temperature programmed oxidation(TPO)from room temperature to 800°C with a rate of 10°C·min-1under a O2/N2mixture(volume ratio,20:80)flow of 100 ml·min-1.
XPS analysis was performed on ESCALAB 250Xi(Thermo Fisher,Germany)operated in the CAE mode using Al Kα radiation as excitation source.The X-ray gun was operated at 210 W power(14 kV,15 mA).The pass energy was set to 30 eV with the step size of 50 meV.
Raman spectroscopy of catalyst samples at various reforming times was performed on in Via(Renishaw,UK)at a wave length of 532 nm for verifying the types of graphite.
2.3.Catalytic reaction
CH4–CO2reforming was carried out in a fluidized bed quartz reactor with an inner diameter of 14 mm at atmospheric pressure.Prior to the reforming reaction,the catalyst was reduced in-situ in H2at a total flow rate of 300 ml·min-1at 800°C for 1 h.The CH4–CO2reforming was performed at 700°C for 12 h.The catalyst loaded was 0.3 g and the gas flow rate was 300 ml·min-1with a CH4/CO2/N2molar ratio of 3:3:4.It should be noted that the Co/MgO catalyst showed good fluidization quality during the reforming reaction attributed to the size and density of the Co/MgO catalyst particles,which was assigned to Geldart-A group.Therefore the effect of defluidization on catalyst deactivation could be ignored.After the reforming reaction,the production gas was analyzed using a micro gas chromatographer(M3000).An ice-cold trap was set between the reactor exit and the gas sampling valve to remove the water formed during the reaction.The conversions of CO2and CH4and selectivity of H2and CO were calculated as follows:


where x,s,F and Ciare the conversion,the selectivity,the gas flow rate and the molar fraction of the i species in the feed/production gas,respectively.The carbon balance across the reactor was within 5%.
In order to investigate the metal sintering and carbon deposition behaviors at various stages of the reforming process,the reforming reaction was interrupted at 1,2,3,6,9,and 12 h.Carbon deposition,Co crystalline structure,and surface valence state of catalyst samples were characterized to show the changes of the catalyst as well as its effect on the activity and stability of the catalysts.
3.Results and Discussion
3.1.Catalytic activity test
Fig.1 shows the conversions of CH4and CO2as a function of time on stream.The initial conversions of CH4and CO2were 68.0%and 74.4%,respectively.The CH4conversion showed an obvious decline to 63%in the first 3 h,then gradually decreased to 58%,while the CO2conversion decreased slightly from 74.4%to 71.5%after reforming reaction for 12 h.Note that the CO2conversion was higher than the conversion of CH4and was not sensitive to reaction time.As shown in Fig.1,the initial H2/CO ratio was approximately 0.93,which decreased slowly to 0.85 after 12 h of reforming reaction.These observations indicated that the reforming reaction was followed by several secondary processes,including the hydrogenation of CO,CO2and the water gas shift reaction(CO2+H2=CO+H2O)[29].

Fig.1.Conversions of CH4 and CO2 and H2/CO ratio as a function of time on stream.
3.2.Catalyst characterization
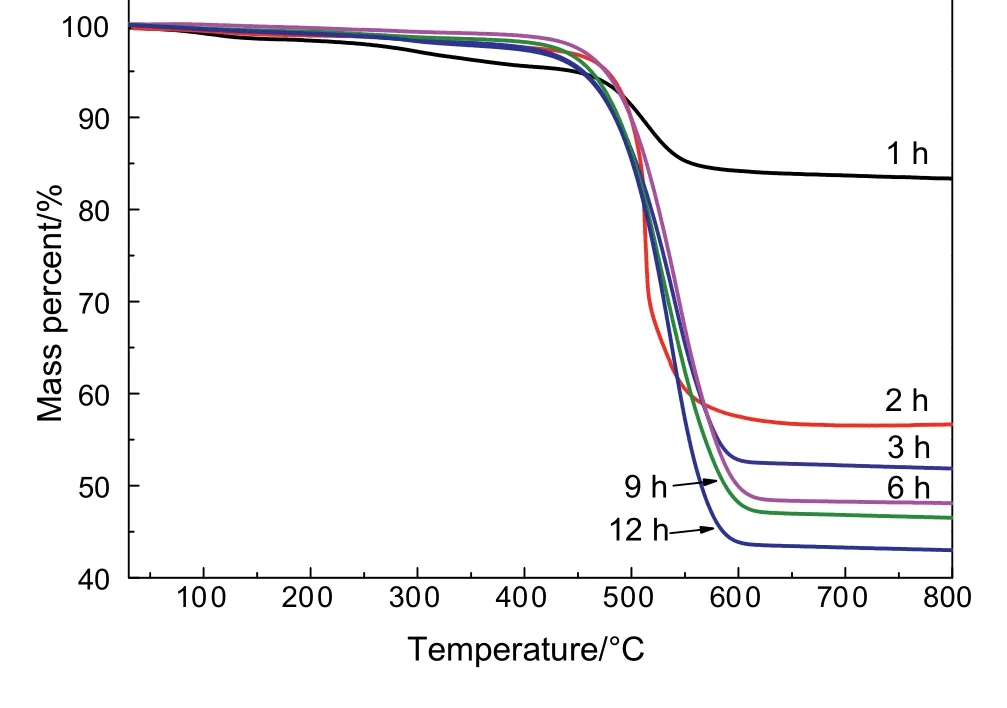
Fig.2.TG curves of the spent catalysts after various times of reforming reaction.
Fig.2 shows the mass loss values of the spent catalysts at various reforming times.It should be mentioned that no obvious mass gain peaks that assigned to the oxidation of the metallic Co around 450°C were observed during the TPO process,indicating a little formation of CoOx[30].In this case,the oxidization of metallic cobalt could be ignored and the mass loss above 450°C should be attributed to the oxidation of the deposited carbon.The mass loss of the spent catalyst showed a large increase in the deposited carbon,from 48.3%at 3 h to 57.1%at 12 h.These results indicated that the carbon deposition was mainly formed in the initial 3 h.Combining Figs.1 and 2,the deactivation of Co/MgO catalyst was attributed to carbon deposition.Previous studies have demonstrated that CH4decomposition and CO disproportion reactions are the two main reactions for carbon deposition[31–33].CO disproportion dominates at lower temperatures,while it is suppressed when the reaction temperature is higher than 700°C,and CH4decomposition reaction is the main reason for carbon deposition at higher temperature ranges[34].Many literatures have reported the carbon deposition amount of Co-based catalysts on various supports such as Al2O3.Compared to the Co/Al2O3catalyst[35],the Co/MgO catalyst in this work displayed better anti-carbon performance and activity under higher GHSV.
Fig.3 shows the Raman spectroscopy of the spent catalysts at various reforming times.The observed Raman shifts around 1330 cm-1,1580 cm-1and 2720 cm-1were assigned to the D,G and 2D bands respectively.The peaks of G and 2D bands indicated the crystal graphite structure which was the characteristic peaks for carbon nanotubes(CNTs)while the peak of D band represented disordered carbon.Therefore,the ID/IGratio of the Raman shift peaks could be related to the extent of filamentous carbon.Fig.3 shows that the ratio of D and G bands changed with the reaction time.In the first 2 h,the characteristic peaks attributed to CNTs were observed with low ID/IGratio(ID/IG=0.62)indicating the formation of ordered graphite,which was compatible with previous reports[36].However,when the reaction time exceeded 3 h,higher ID/IGratio(ID/IG=1.55)was observed,indicating the increase of disordered coke.This was contradictory with the previous findings[37].We may assume that there exists another mechanism for such carbon deposition which would be discussed later.

Fig.3.Raman shift of the spent catalysts at various reforming times.

Fig.4.TEM images of reduced and spent catalysts.(a)Reduced,(a′)enlarged image of region in(a),(b)used for 1 h,(b′)enlarged image of region in(b),(c)used for 2 h,(c′)enlarged image of region in(c),(d–g)used for 3,6,9,and 12 h respectively.
Fig.4 shows the TEM images of the fresh and spent catalysts.The fresh Co/MgO catalyst exhibited a low BET area(22 m2·g-1)with the average Co NP size of 8.1 nm dispersed on the external surface of MgO flake(Fig.4(a)).After the reforming reaction,no obvious sintering of Co NPs was detected in all spent cases.However,two types of carbon deposition were observed in the spent catalysts(Fig.4(b)–(g)).One was CNTs which grew on the tip of Co NPs,while the other one was amorphous carbon or coke that coated on the surface of Co NPs,which in some literatures was named as carbon nano-onions(CNOs)[37–39].Many researches have confirmed that CNTs would not totally deactivate the active metal center unless the whole external surface of carrier was blocked.For example,Li[38]and Zhou[39]reported similar results that Fe0NPs dispersed in the pore of zeolite crystallites deactivated due to the blockage of carbon,while those on the external surface and anchored on the tip of CNTs were still available for catalytic methane decomposition.

Fig.5.XRD patterns of calcined,reduced and spent catalysts and enlarged view of 42°–44°.

Table 1 EDS spectra of the spent catalyst as shown in Fig.4(atom%)
After the reforming reaction for 1 h,several CNTs were observed (Fig. 4(b)), which agreed with the Raman results.Disordered graphite with 5–6 layers coated on the surface of Co NPs was also found in Fig.4(b′).The layer numbers of the multilayered graphite increased to more than 10 layers after 2 h of reaction(Fig.4(c)and(c′)).However,the layer numbers would not increase further with reaction time(Fig.5(d)–(g)).For this type of carbon deposition,Co NPs were totally coated within the layers of CNOs,which would cover the active metal center.Hence,this inhibited a further increase of carbon layer numbers.EDS spectra of two regions with different quantity of layers were shown in Table 1.Much more O was detected on the surface of the Co NPs with more layers of carbon(generally,increased from 0.03%to 39.29%).Generally speaking,there will be more errors about the carbon layer number and O content on the surface of the Co NPs by TEM/EDS analysis of only two particles.However,these results reflected the relationship between metal oxidization and morphology of deposit carbon.It suggested that the existence of O might have an effect on the morphology of coke.In order to illustrate the different formation behaviors of CNTs and CNOs,crystalline structure and surface valence state of catalyst were investigated by XRD and XPS methods.
Fig.5 gives the XRD patterns of fresh and spent catalysts.The peak of MgO at 42.7°shifted to a higher degree,indicating the formation of solid solution[22].The diffraction peak around 26°that assigned to deposited carbon showed puny difference with the increase of reaction time,which was in accordance with the TG results(Fig.2).The peak near 44°of the spent catalysts after reforming reaction showed no obvious increase as compared to the reduced catalyst,indicating a strong force between metal NPs and support and high dispersion due to the formation of CoMgOxsolid solution[36].The data of crystalline size calculated using the Scherrer equation were listed in Table 2.It should be noticed that the initial Co particle size of the newly reduced catalyst was 13 nm and changed slightly to 8.1 nm after reaction for 12 h,which was similar with the results from TEM.To our knowledge,for Ni-and Co-based catalysts,sintering of metal particle under high temperature was the main reason for carbon deposition.However in this research particles changed slightly due to the strong interaction of solid solution and we could confirm that sintering was not the reason for carbon deposition.
The Co 2p XPS spectra of fresh and spent Co/MgO catalysts are shown in Fig.6.The CoO/MgO before reduction showed a strong peak at 780.3 eV with a satellite peak at nearly 787.0 eV,which had a large peak shift to higher binding energy compared to standard Co 2p3/2of Co3O4at 779.5 eV[40].This is attributed to the formation of CoMgOxsolid solution,which was confirmed by the XRD results(Fig.5).The satellite peaks indicated different chemical environments of Co species corresponding to the force with carrier.Chen et al.[41]found the strong interaction force of CoAl2O4caused Co 2p3/2peak shifting to high binding energy,from 779.5 eV to 781.9 eV,which was compatible with our results.

Table 2 Physicochemical properties of the calcined,reduced and spent Co/MgO catalysts after reforming reaction at various times

Fig.6.XPS spectra of calcined,reduced and spent catalysts.
After reduction,a distinct peak appeared at the low binding energy side at 778.3 eV attributed to the metallic cobalt[40].Meanwhile,the peak intensity at 780 eV and 787 eV was enhanced,indicating Co oxidization species penetrated further to MgO lattice.After the reforming reaction for 3 h,the peak for metallic Co at 778.3 eV was equivocally detectable.In addition,an obvious peak at 779.5 eV corresponding to Co3O4was observed after the reforming reaction.Table 2 shows that the ratio of metallic Co on the surface of catalyst to the whole Co(Co0/Cototal)determined by XPS decreased with the increasing reaction time especially in the initial 3 h.This indicated that the metallic cobalt on the surface was oxidized to oxidization species during reaction resulting in a decrease of metal content on the surface.

Fig.7.H2-TPR measurement of calcined,reduced and spent catalysts.
It should be mentioned that only a thickness of 5 nm of catalyst surface could be detected by XPS and part of metal particle might be shielded by deposit carbon.In order to measure the amount of Co oxidization species,H2-TPR experiments were taken for the listed catalysts respectively.Fig.7 gives the H2-TPR profile of fresh and spent catalysts.It has been reported that the TPR peaks at 200°C,400°C and 1000°C were assigned to three types of Co oxidization species,corresponding to an increasing interaction force with MgO support[21,42].In our study,before reduction,three types of O2-were detected at nearly 330°C,450°C and 900°C,representing the individual Co3O4on the surface[42],the Co oxidization species in deeper lattice and ideal CoMgOxsolid solution[21]respectively.The higher reduction temperature indicates the stronger force between the Co NPs and the support.The formation of CoMgOxsolid solution played an important role in maintaining its strong metal–support interactions and stability of the active metal size,as evidenced from the H2-TPR characterization.After the reduction for 1 h,there was still part of CoOxleft with a detectable peak at 380°C.After the reforming reaction,the reduction peak was shifted to above 581°C for the spent catalyst samples.This is attributed to cobalt oxidization species trapped in deeper lattice of MgO due to higher reaction temperature.After the reforming reaction,the intensity of peaks around 330°C increased with the reaction time,indicating the newly generated Co3O4.The H2consumption was listed in Table 2.Fig.8 shows carbon deposition and H2consumption as a function of time on stream.Both carbon deposition and H2consumption displayed almost the same trends:a rapid increase in the initial 3 h,and followed by a steady increase in the next 9 h.Combining the results of EDS and H2-TPR,we can draw a conclusion that the significant carbon deposition is related to the oxidization of Co NPs.

Fig.8.Carbon deposition and H2 consumption as a function of time on stream.
The morphologies and formation mechanism of deposited carbon from methane catalytic decomposition have been previously reported.In the literatures,there exist two parallel processes on the surface of transition metal namely carbon deposition and carbon diffusion.The morphologies of accumulated carbon are determined by the rate of the above two processes.CNOs form if the deposition rate is higher than diffusion rate,otherwise CNTs or MWNTs form[39,43].Zhou[43]confirms the diffusion rate decreases with metal size and there is a strong tendency to form CNOs on Ni particles above 70 nm.In this paper,both CNOs and CNTs were detected,although the size of Co NPs was small and showed little change during CH4–CO2reforming.

Fig.9.Carbon deposition mechanism.
Here we propose a possible mechanism model for the formation of CNTs and CNOs on the Co/MgO catalyst.As is shown in Fig.9,methane was firstly decomposed on the metal surface,generating CHxspecies and adsorbed hydrogen[44,45].CO2from gas phase was absorbed on metal surface and dissociated to CO and oxygen species(OH intermediates)with the effect of active hydrogen.Then CHxspecies were eliminated by reacting with OH generating CO and hydrogen.From this mechanism,the oxygen species generated from CO2dissociation plays a key role in estimating CHxspecies.With the participation of oxygen species,a portion of carbon was eliminated in time.Therefore,carbon diffusion rate was much higher than carbon accumulation rate.As a result,ordered CNTs with high extent of graphite were formed(as shown in Fig.9,PATHWAY 1).In this research,significant carbon deposition with low extent of graphite occurred with the appearance of metal oxidization.Hence,we can draw a conclusion that the formation of CNOs was attributed to the oxidization of metal NPs.As shown in Fig.9,PATHWAY 2,in the preliminary stage of reaction,O species originated from dissociated CO2had a strong tendency to combine with metal NPs rather than CHxspecies so that layered carbon accumulated around Co particles and its rate was much faster than the diffusion rate.Therefore,CNOs were deposited after the graphitization process under high temperature.The atmosphere was both reductive and oxidative for the reductive(CH4,H2and CO)and oxidative(CO2and H2O)species that coexisted in the reactor.During the reaction a fraction of Co0was oxidized to CoOxspecies which was reduced again to Co0later.The oxygen transfer was accomplished by a dynamic redox process.However the oxygen transfer may be interrupted through generating CoMgOxwhich was difficult to reduce.However,in this research only a part of Co particles were oxidized and coated in CNOs while the others formed CNTs.This Co NP oxidation phenomenon is not clear now,and needs to be investigated in the future.One of the possible controlling factors is the interaction between active metal particles and nearby support.It has been proved that the metal–support interaction has a significant effect on the reaction progress of CH4–CO2reforming[4].This metal–support interaction might contribute to different degrees of the Co oxidization.The Co NPs that are close to the MgO support are exposed to more oxidative intermediate generated from CO2dissociated adsorption,and those are easy to be oxidized to form CoOx.However,the Co NPs away from the support are not easy to be oxidized due to the lower reactant concentration.This indicates that although the high dispersion of metal particles leads to advanced performance of reaction activity,the strong internal force between the support and metal could promote Co metal oxidization.This opinion makes sense on why metal oxidization is only detected in low metal loading samples in the literatures[29,46].
4.Conclusions
Deactivation and carbon deposition behaviors of a Co/MgO catalyst for CH4–CO2reforming in various reaction times were investigated.No obvious sintering of Co nanoparticles(NPs)was observed during the reforming reaction due to the formation of CoMgOxsolid solution.Significant carbon deposition was observed throughout the reforming reaction.Rapid deactivation occurred mainly in the first 3 h due to carbon deposition in the form of carbon nano-onions(CNOs),which coated and blocked the Co NPs.This severe carbon deposition was originated from the oxidation of Co NPs.The Co NPs had a strong tendency to combine with oxidization species,where carbon species would accumulate to form CNOs and block the active centers and lead to catalyst deactivation.
杂志排行

Chinese Journal of Chemical Engineering的其它文章
- Ni/bentonite catalysts prepared by solution combustion method for CO2 methanation☆
- Modelling of a post-combustion carbon dioxide capture absorber using potassium carbonate solvent in Aspen Custom Modeller
- Mass transfer correlations for membrane gas-solvent contactors undergoing carbon dioxide desorption
- The CO2 absorption and desorption performance of the triethylenetetramine+N,N-diethylethanolamine+H2O system☆
- Recent developments and consideration issues in solid adsorbents for CO2 capture from flue gas☆
- High-efficiency and pollution-controlling in-situ gasification chemical looping combustion system by using CO2 instead of steam as gasification agent