基于信息熵的加权基因关联网络融合方法
2018-03-26伍度志
伍度志,杨 帆,赵 静
(1.重庆工商大学融智学院 重庆 巴南区 401320;2.陆军勤务学院数学教研室 重庆 沙坪坝区 401331;3.上海中医药大学交叉科学研究院 上海 浦东新区 201203)
加权基因关联网络(weighted gene association network, WGAN)是表示基因间功能相关关系的复杂网络[1],其中节点代表基因,边代表基因间的相互作用,权重代表相互作用的可信度。WGAN网络的构建是为了克服目前已有的生物学实验数据与实际存在的基因功能相关关系相比严重不足、以及高通量实验的结果存在严重噪声的问题。通常采用计算方法整合与基因的功能联系相关的各种生物学特征的数据源,推断基因之间的关联关系,并对每一对关联关系赋予置信分,作为网络中边的权重,从而构建加权的基因关联网络。因此这类网络既包含了一些特定类型的基因或蛋白间的相互作用信息,如蛋白-蛋白相互作用[2](PPI)、基因共表达[3]、转录调控[4]、信号通路[5]等,又比特定类型的分子网络包含更广泛的信息。
目前,基因相关关系的数据融合方法主要分为主观打分融合方法、相似性融合方法和统计推断打分方法3种类型。通过这些方法已经构建了一些WGAN网络,就人类基因组而言,有HIPPIE[6]、HumanNet[7]、STRING[8]以及FunCoup[9]网络等。文献[6]收集了现有的蛋白-蛋白相互作用数据库BioGrid[10]、IntAct[11]、MINT[12]、DIP[13]、BIND[14]等中的数据,基于试验方法的先进性、支持基因间关联关系的文献数目以及在非人类物种中存在该连接的基因对数目3种不同的信息,自定义了一种基因对的打分方法,对每一对基因间的关联关系的可靠性进行打分,从而构建了HIPPIE网络。文献[7]基于概率似然比提出一种统一的网络边权打分方法,该方法以基因本体注释数据库GO(gene ontology)[15]为背景网络,对21个基因功能数据集中的每一条边进行重新打分,得到了HumanNet网络。文献[8]通过建立朴素贝叶斯分类器模型方法,融合多种与基因关联关系相关的生物学数据源,得到了一个加权基因关联网络STRING网络。文献[9]选取了八大真核生物体的大规模数据,通过朴素贝叶斯模型方法融合得到了FunCoup网络。
基于网络的复杂疾病病理学和药理学的研究,广泛应用人类全基因组加权基因关联网络作为背景网络,以识别疾病相关基因、探测药物对应的网络药靶,从而加深复杂疾病的医学认识、改进复杂疾病的治疗。可以想见,背景网络的质量,与相关研究结果的精确度是相关的。现有的人类全基因组基因关联网络如HumanNet、STRING和FunCoup等,各自在生物学基础研究及疾病研究中都有成功应用的案例[16-18]。然而,这些网络间却存在着巨大差异。它们虽然包含了80%以上相同的基因,但拥有的相同的关联边却很少,低于各自总边数的10%。如果在这些已有的WGAN的基础上,进一步识别其中包含的正确信息,将它们融合成一个信息更全、更准确的加权基因关联网络,对于更好地从系统水平理解细胞内部生物学过程、以及研究复杂疾病的病理,都是很有意义的。
本文利用信息熵[19]刻画基因连边权重的不确定度,提出了基于信息熵理论的融合策略,在现有4个人类全基因组WGAN基础上,充分利用多个网络中所有连边的信息来构造包含更多节点和边的WGAN。本文将原有网络及新构建的网络分别用于肥胖症的疾病基因预测[20],以检验新网络的应用价值。
1 WGAN网络融合模型
1.1 网络边权的不确定度刻画
熵是衡量某一个体系混乱程度的变量,它在不同领域被引申为更为具体的解释。在研究随机现象的过程中,熵用来描述随机现象发生的平均不确定度,为评估随机现象发生的不确定程度提供了一个定量的指标。同样,这一指标也被广泛应用于信息理论的研究领域,被称为信息熵。
对于某一随机现象X,若X包含n种可能的结果,且分布率为则随机现象X发生的不确定程度可以通过信息熵定义如下:

本文拟将此方法应用于WGAN网络的融合。对于WGAN网络,可以通过适当的归一化方法,使它的边权取之于区间(0,1]。因此,在后面的描述中,总假设WGAN网络中的边权取之于区间(0,1]。 假设现有m个WGAN网络N1,N2,…,Nm,它们具有相同的基因,其中网络Nk中i、j基因节点的连边权重记为则融合这m个WGAN网络就是要将网络中任意基因对i、j的连边权重融合成一个新的权重,作为融合后网络中基因对i、j的连边权重W(ij)。由于现有的融合算法主要限于线性融合,因此,上面的融合问题转化为寻找融合系数使:
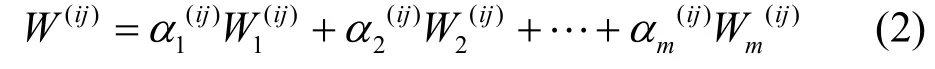
为了寻找合理的融合系数,需要对每一组基因对连边进行深入分析。由于WGAN网络中的边权取之于区间(0,1],因此,W(ij)可以理解为WGAN网络中基因i、j连边的概率,由此可以定义如下随机现象Y:

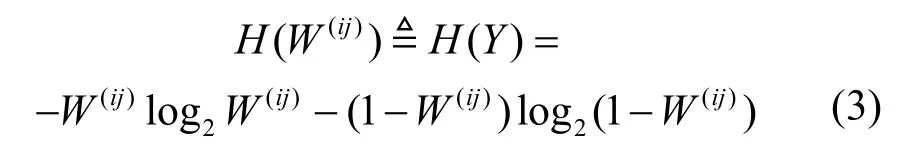
因此,WGAN网络中基因i、j连边的不确定程度可以通过式(3)来刻画。显然,式(2)中融合系数的设计与连边自身的不确定程度密切相关,这为融合系数设计提供了有价值的途径。
1.2 WGAN网络连边权重预处理
在实际情况中,同一对基因可能在一些网络中存在连边,而在另一些网络中不存在连边,对于后者,用式(3)来刻画其连边的不确定性显然是不合适的,因为式(3)中要求连边概率W(ij)大于零。为了处理这种情况,需要对基因对的连边做适当的处理,从而使得融合更加合理。本文先求得背景网络的连边并集N,则N中的每一条连边都对应着h个权重且h≤m,对于那些在一些网络中存在连边,而在另一些网络中不存在连边的基因对,假设其在对应背景网络上也存在连边并将其权重设为一个非常小的数值ε。通过这种处理,N中每一条连边都存在m个权值,从而可以利用式(3)来设计融合系数。同时,如果网络的一组基因对的连边权重为1,则重新修改它的权重为1−ε。从而,通过预处理后的各WGAN网络中的基因对连边的最小权值为ε。
1.3 基于信息熵的WGAN网络数据融合模型
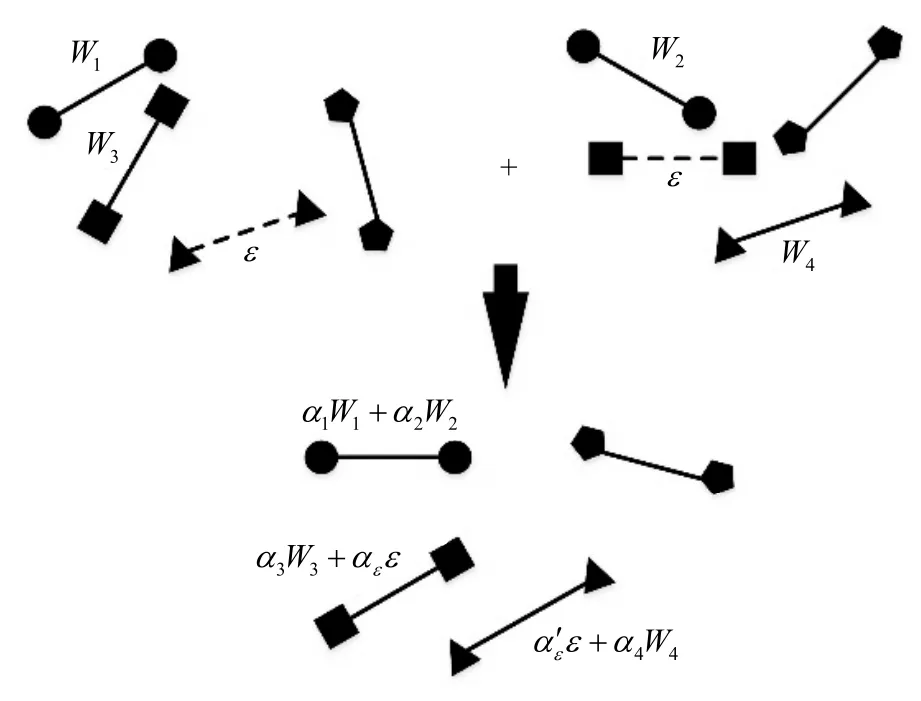
图1 网络融合过程简略图
对第k个WGAN网络Nk的每一组基因对i、j的连边权重利用式(3),可以定义该连边的不确定程度越大,则该连边的不确定程度越大,因此,在确定融合系数时,应该赋以相应连边的融合系数一个比较小的值,反之则赋以一个比较大的融合系数。为此,对各网络的每一组基因对i、j的连边,引入如下函数:

式中,θ>0为调整因子,主要用于调整基因对i、j连边的不确定程度对函数的影响程度。在实际应用中,可以通过训练的方法来选择适当的参数θ(见1.4节)。不难分析出,函数是基因对i、j连边的不确定程度单调递减函数,因此可以利用它来定义相应的融合系数。通过对函数做归一化处理,定义m个WGAN网络中基因对i、j连边的融合系数为:
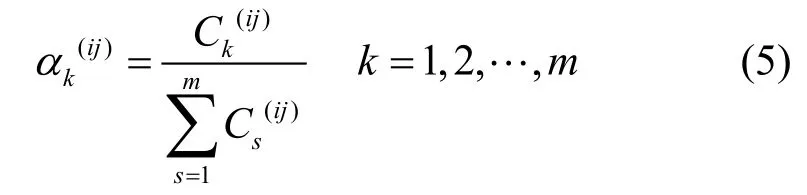
则对这m个WGAN网络融合后的网络中基因对i、j连边的连边权重为。图1为两个网络的融合过程简略图。
1.4 模型参数确定
根据基因本体注释数据库GO中全体人类基因的功能信息,构建GO网络,并将它作为测试网络,确定融合模型的参数。GO数据库是基因本体联合会(Gene Onotology Consortium)所建立的数据库,该数据库对大量物种中的每个基因和蛋白质的功能用标准的生物学词汇条目(GO term)进行描述。本文构建的GO网络中节点代表人类基因,若两个基因至少有一个共同的GO term, 则它们对应的节点有连边,连边的权重为这两个基因共有的GO term的数目,并将其归一化到(0,1]区间。因此GO网络中基因的连边代表两个基因在生物功能上有相关性,边权则代表这个相关性的强弱程度。
θ为调节信息熵对融合系数影响程度的调整因子。为了选择较为合适的调整区间,本文通过分析式(4)的函数模型,分别选取不同的θ(0~5,以0.1为步长)以及不同的信息熵值H(0.05~1,以0.05为步长)作为自变量,观测比较了θ和H对融合系数的影响程度,如图2所示。
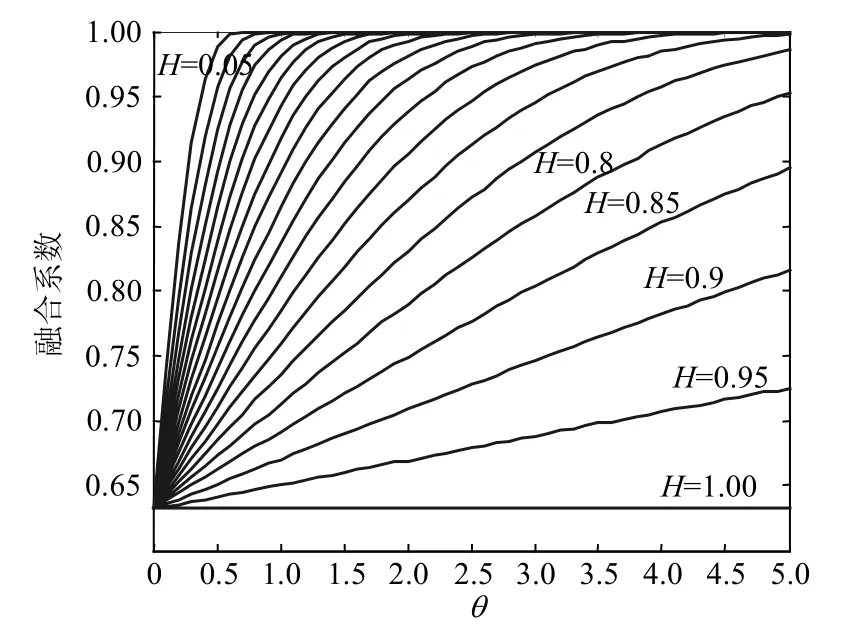
图2 θ和H对融合系数的影响
由图2可以看出,当边权的信息熵值H比较小时,θ只有取值略小,才能使融合系数具有有效的区分度;当边权的信息熵值H比较大时,θ只有取值稍大,才能使融合系数具有有效的区分度;由于加权基因网络的边权信息熵值大小分布不均,为了使融合系数都具有有效的区分度,因此建议选择θ的调整区间为(0,3)。
本文将把融合后网络的权值与GO网络的权值进行比较分析,计算其共同连边权值对应的差平方和,并且在这个值达到最小时选取对应的模型参数θ,从而将模型参数的确定转化为优化的求解问题:

式中,Wθ表示参数条件下的融合后网络连边权值;WGONet表示GO网络连边权值。
2 融合算法实例
2.1 原始网络预处理
本文针对提出的网络融合模型,对4个现有的人类全基因组加权基因关联网络,即HIPPIE、HumanNet、FunCoup和STRING进行融合。将这4个原始WGAN网络分别记为4个网络的基因数和连边信息如表1所示。

表1 4个原始网络的基本信息
因为4个网络的连边信息和节点信息各不相同,因此首先需要按照1.2节中的方法处理。先求得4个基因网络的并集网络N,再按照1.2节中的方法来补充定义某些基因对的连边权重。经过这种处理,4个网络的每一条连边都对应着4个权值,即分别为4个子网络所对应的权值。截取并集网络的一部分表2所示。
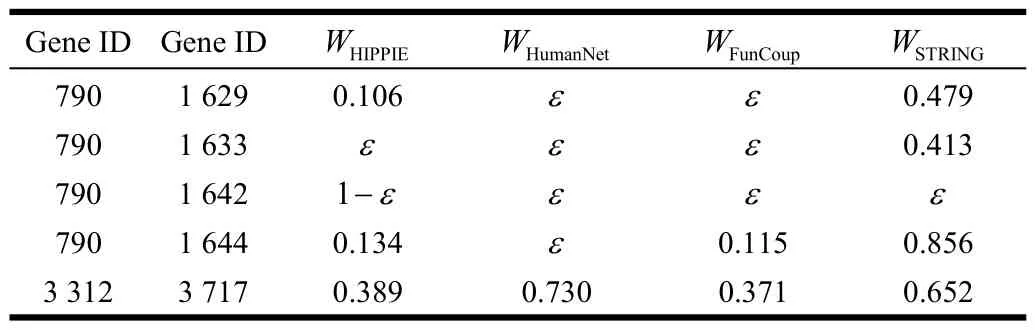
表2 并集网络N的部分数据
表2中,Gene ID表示某个基因的Entrez ID,W表示各背景网络中对应边的权重。本文研究中,取ε=0.001。
2.2 4个WGAN网络的融合
根据式(4)、式(5),需要计算各网络中每一组基因对i、j连边的函数和融合系数为了选取比较合适的调整因子,本文选取了GO网络作为训练网络,分步长对融合系数函数中的θ参数进行训练。通过比较分析,实验结果得到的θ和f(θ)变化关系如图3所示。
由图3可以看出,f(θ)随θ的变化先呈现递减后递增的趋势,在θ取0.3时,f(θ)达到最小。
在取θ为0.3的情况下,利用式(4)、式(5),可以将4个网络每一组基因对的i、j连边进行融合,从而得到一个新的网络FN, 其节点数为19 490,边数为7 092 510。
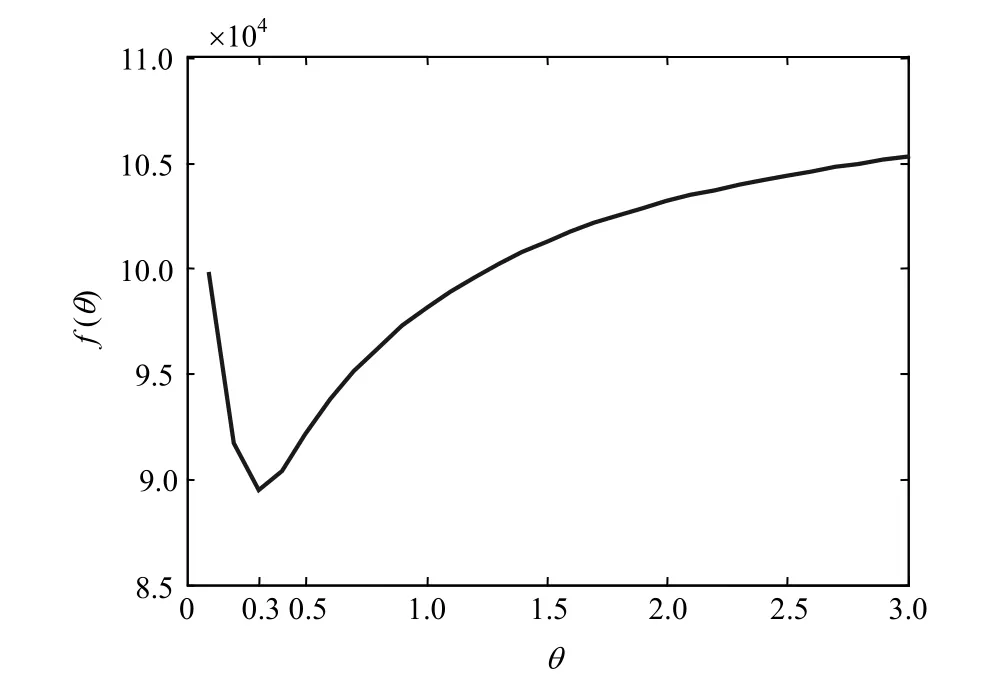
图3 参数θ的训练
然后,比较融合前后网络与GO网络的共同连边数以及共同连边权重的Person相关系数,得到图4。
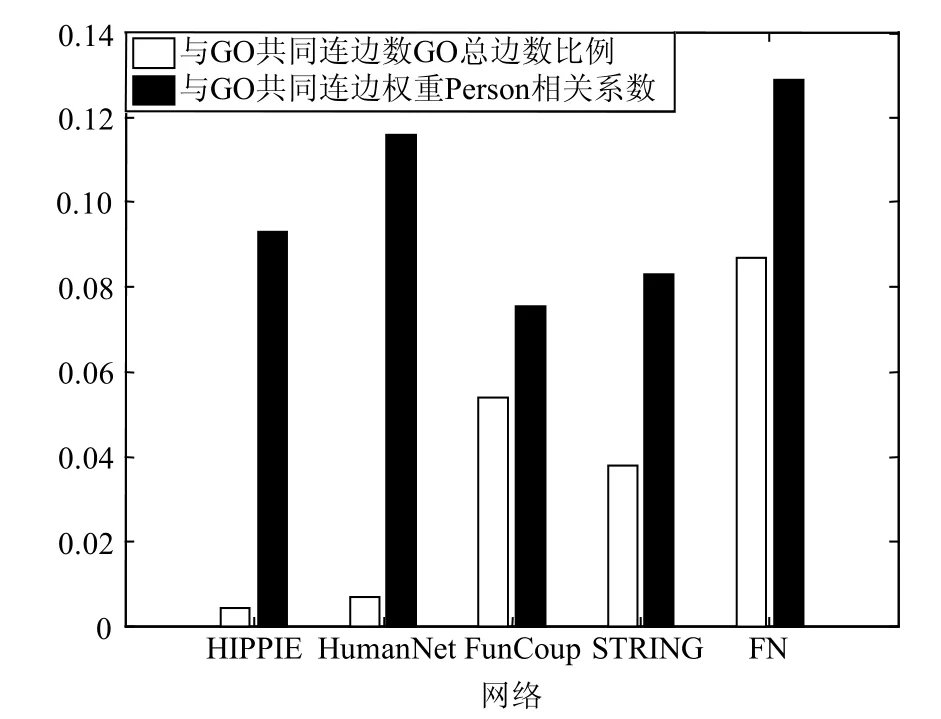
图4 融合前后网络与GO网络的比较
从图4可以看出,相比原始网络,融合后的网络FN连边信息更加丰富,并且其权重经过融合后,与GO共同连边权重的Person相关系数相比原始网络有显著提高,说明FN的边权比原始网络的边权有更强的生物学相关性。
3 肥胖症的疾病基因预测
生物系统是由多分子和基因相互作用的结果。复杂疾病的基因不是孤立存在的,基因与基因之间有相互作用。加权基因关联网络的研究为系统生物学和疾病分子预测提供了一个崭新的平台,对预测疾病相关基因做出了较大的贡献。由于相同的疾病基因在基因关联网络中具有邻近性,因此基于网络的预测方法被广泛应用于疾病基因预测研究中。为了检验融合后网络的实用性,本文分别将融合前后的网络作为背景网络,进行肥胖症的疾病基因预测。
3.1 预测方法概述
基于网络的疾病基因预测方法将已知的疾病基因作为先验信息组成种子集,根据候选基因与种子基因在网络上的拓扑关系,预测候选基因是疾病基因的可能性。本文采用直接邻居法[21],该方法是把与已知疾病致病基因直接相连的基因作为疾病的可能致病基因,基于全网络对每一个基因进行打分,得出其与已知致病基因直接相连的总得分Si,即与致病基因直接相连的边的权重总和。其模型为:

式中,Wij表示基因i和致病基因j的连边权重;seed表示已知致病基因集。由此可得全网络中的每一个基因的得分值,再将所有基因依据其分值由大到小进行排序。本文截断出排名前n个基因,计算预测准确值,即测试集中的基因在这n个基因中所占的比例。
本文从人类孟德尔遗传在线数据库OMIM(online Mendelian inheritance in man, OMIM)[22]和文献中收集已知的肥胖症(obesity)的致病基因。其中从OMIM数据库获得24个肥胖症致病基因,从文献[23]中获得与肥胖症相关的373个基因。
3.2 预测效果分析
本文用两种方法检验疾病基因的预测效果,一种是留一交互验证法[24], 另一种是模拟寻找疾病基因的方法[21]。

图5 用留一交叉验证比较融合前后网络的疾病基因预测效果
在留一交叉验证法中,将OMIM中的24个疾病基因与文献中的373个疾病基因合并,得到已知的肥胖症疾病基因集合。每次利用此集合中的一个疾病基因构成测试集,剩余的疾病基因构成种子集。用式(7)对背景网络中的每个基因打分,验证算法是否能够成功地预测测试基因为致病基因。图5显示了按分值排名截取不同比例的网络基因组总基因数时,以不同网络为背景网络所得到的预测准确率,即在不同的比例下,合并疾病基因集中被预测到的疾病基因占集合总基因数的比值。可以看出,融合后的网络FN与网络STRING取得了最好的预测准确率。
在模拟寻找疾病基因的方法中,本文以OMIM中的24个疾病基因构成种子集,文献[23]中的373个疾病相关基因作为测试集,对网络进行打分。分别选择得分最高的20和200个基因为预测的疾病基因,比较融合前后背景网络下被预测到的疾病相关基因所占比例。以排名的截断值为横坐标,以预测准确值为纵坐标将融合前的4个网络HIPPIE、HumanNet、FunCoup、STRING和融合后的网络FN做疾病基因预测效果对比分析,如图6所示。

图6 融合前后的网络预测效果比较
由图6可以看出,当截断值为20时,融合后的网络FN的预测准确值比4个原始网络高;当截断值为200时,融合后的网络FN的预测准确值和STRING相当,显著高于其他3个网络的预测效果。
这些结果说明,本文融合后的网络FN可以成功地用于疾病基因预测。
4 结束语
本文研究是加权基因关联网络数据融合方面的一个新的尝试,提出了一种基于信息熵的WGAN网络数据融合方法,将现有的人类加权基因关联网络的信息进行整合。通过此方法,本文构建了一个包含现有网络所有节点和边信息的融合网络FN。通过与GO网络对比显示,FN的边权比原始网络中的边权有更强的生物学相关性。将FN与原始网络同时用于肥胖症的疾病基因预测,发现FN的预测效果高于或相当于效果最好的原始网络STRING,说明此网络可以用于疾病基因预测。此工作在生物网络数据整合以及疾病基因预测的研究方面都有重要的价值。
[1]周涛, 张子柯, 陈关荣, 等.复杂网络研究的机遇与挑战[J].电子科技大学学报, 2014, 43(1): 1-5.ZHOU Tao, ZHANG Zi-ke, CHEN Guan-rong, et al.The opportunities and challenges of complex network research[J].Journal of University of Electronic Science and Technology of China, 2014, 43(1): 1-5.
[2]WILLIAMSON M P, SUTCLIFFE M J.Protein-protein interactions[J].Biochemical Society Transactions, 2010,38(4): 875-878.
[3]ZHANG B, HORVATH S.A general framework for weighted gene co-expression network analysis[J].Statistical Applications in Genetics and Molecular Biology, 2005, 4(1):1128.
[4]CILIBERTO G, COLANTUONI V, DE FRANCESCO R, et al.Transcriptional control of gene expression in hepatic cells[M]//KARIN M.Gene Eexpression: General and Cell-Type-Specific.[S.l.]: Birkhäuser, 1993.
[5]MARTINI P, SALES G, MASSA M S, et al.Along signal paths: an empirical gene set approach exploiting pathway topology[J].Nucleic Acids Research, 2013, 41(1): e19.
[6]SCHAEFER M H, FONTAINE J F, VINAYAGAM A, et al.HIPPIE: Integrating protein interaction networks with experiment based quality scores[J].PloS One, 2012, 7(2):e31826.
[7]LEE I, BLOM U M, WANG P I, et al.Prioritizing candidate disease genes by network-based boosting of genome-wide association data[J].Genome Research, 2011, 21(7):1109-1121.
[8]FRANCESCHINI A, SZKLARCZYK D, FRANKILD S, et al.STRING v9.1: Protein-protein interaction networks, with increased coverage and integration[J].Nucleic Acids Research, 2013, 41(D1): D808-D815.
[9]ALEXEYENKO A, SONNHAMMER E L.Global networks of functional coupling in eukaryotes from comprehensive data integration[J].Genome Research, 2009, 19(6): 1107-1116.
[10]CHATR-ARYAMONTRI A, BREITKREUTZ B J,OUGHTRED R, et al.The BioGRID interaction database:2015 update[J].Nucleic Acids Research, 2015, 43(D1):D470-D478.
[11]HERMJAKOB H,MONTECCHI‐PALAZZI L,LEWINGTON C, et al.IntAct: an open source molecular interaction database[J].Nucleic Acids Research, 2004,32(suppl 1): D452-D455.
[12]CHATR-ARYAMONTRI A, CEOL A, PALAZZI L M, et al.MINT: the molecular INTeraction database[J].Nucleic Acids Research, 2007, 35(suppl 1): D572-D574.
[13]XENARIOS I, SALWINSKI L, DUAN X J, et al.DIP, the database of interacting proteins: a research tool for studying cellular networks of protein interactions[J].Nucleic Acids Research, 2002, 30(1): 303-305.
[14]BADER G D, BETEL D, HOGUE C W V.BIND: the biomolecular interaction network database[J].Nucleic Acids Research, 2003, 31(1): 248-250.
[15]Gene Ontology Consortium.The gene ontology (GO)database and informatics resource[J].Nucleic Acids Research, 2004, 32(suppl 1): D258-D261.
[16]RE M, VALENTINI G.Random walking on functional interaction networks to rank genes involved in cancer[C]//IFIP International Conference on Artificial Intelligence Applications and Innovations.Berlin,Heidelberg: Springer, 2012: 66-75.
[17]TABOADA B, VERDE C, MERINO E.High accuracy operon prediction method based on STRING database scores[J].Nucleic Acids Research, 2010, 38(12): e130.
[18]ZHAO J, WANG C L, YANG T H, et al.A comparison of three weighted human gene functional association networks[C]//2012 IEEE 6th International Conference on Systems Biology (ISB).[S.l.]: IEEE, 2012: 26-31.
[19]COVER T M, THOMAS J A.Elements of information theory[M].[S.l.]: John Wiley & Sons, 2012.
[20]吕琳媛.复杂网络链路预测[J].电子科技大学学报,2010, 39(5): 651-661.LÜ Lin-yuan.Link prediction on complex network[J].Journal of University of Electronic Science and Technology of China, 2010, 39(5): 651-661.
[21]LINGHU B, SNITKIN E S, HU Z, et al.Genome-wide prioritization of disease genes and identification of disease-disease associations from an integrated human functional linkage network[J].Genome Biology, 2009,10(9): 1-17.
[22]HAMOSH A, SCOTT A F, AMBERGER J S, et al.Online mendelian inheritance in man (OMIM), a knowledgebase of human genes and genetic disorders[J].Nucleic Acids Research, 2005, 33(suppl 1): D514-D517.
[23]HANCOCK A M, WITONSKY DB, GORDON A S, et al.Adaptations to climate in candidate genes for common metabolic disorders[J].PLoS Genetics, 2008, 4(2): e32.
[24]REFAEILZADEH P, TANG L, LIU H.Crossvalidation[M]//Encyclopedia of Database Systems.[S.l.]:Springer US, 2009: 532-538.
