甘草黄酮类化合物对CDK1的抑制活性及体内外抗肝癌活性
2018-03-20李晶晶
张 敏,李晶晶
(1长江职业学院,武汉 430074;2湖北中医药大学基础医学院,武汉 430065)
甘草来源于豆科植物乌拉尔甘草、胀果甘草或光果甘草的干燥根和根茎,具有清热解毒、补脾益气、润肺、祛痰止咳、调和诸药等功效,是临床上应用最广泛的传统中草药,其主要成分为三萜类和黄酮类化合物[1]。长期以来,对于甘草的有效成分和药理作用研究主要集中在三萜类化合物甘草酸和甘草次酸[2-3]。近年来,现代药理学研究发现甘草中黄酮类化合物也是一类非常有效的活性成分,具有多种药理活性,如抗菌、抗炎、抗溃疡、抗氧化、抗糖尿病、抗肿瘤、抗抑郁等作用[4-5],且毒性低、不良反应少,具有良好的临床开发潜力。最近研究表明,甘草黄酮类化合物对肝癌Bel-7402细胞展现出了较强的抑制活性[6]。因此,本研究继续探讨甘草黄酮单体化合物甘草查耳酮A-F、刺甘草查耳酮、异甘草素、异甘草苷、光甘草定、甘草素、甘草苷的抗肝癌活性,以期得到高效、低毒的抗肝癌先导化合物。
尽管目前对甘草黄酮的抗肿瘤作用靶点研究较少,但是参考黄酮类化合物抗肿瘤作用靶点发现,黄酮类化合物对细胞周期依赖性蛋白激酶(CDK)有较强的亲和力[7],而CDK是控制细胞周期与增殖过程的重要激酶,已成为新型抗肿瘤药物设计的重要靶点,尤其是CDK1/cyclin B在多种肿瘤中过表达[8]。因此,本研究首先测试了12个甘草黄酮(图1)对CDK1/cyclin B的抑制活性,以判断甘草黄酮类化合物的靶向性。
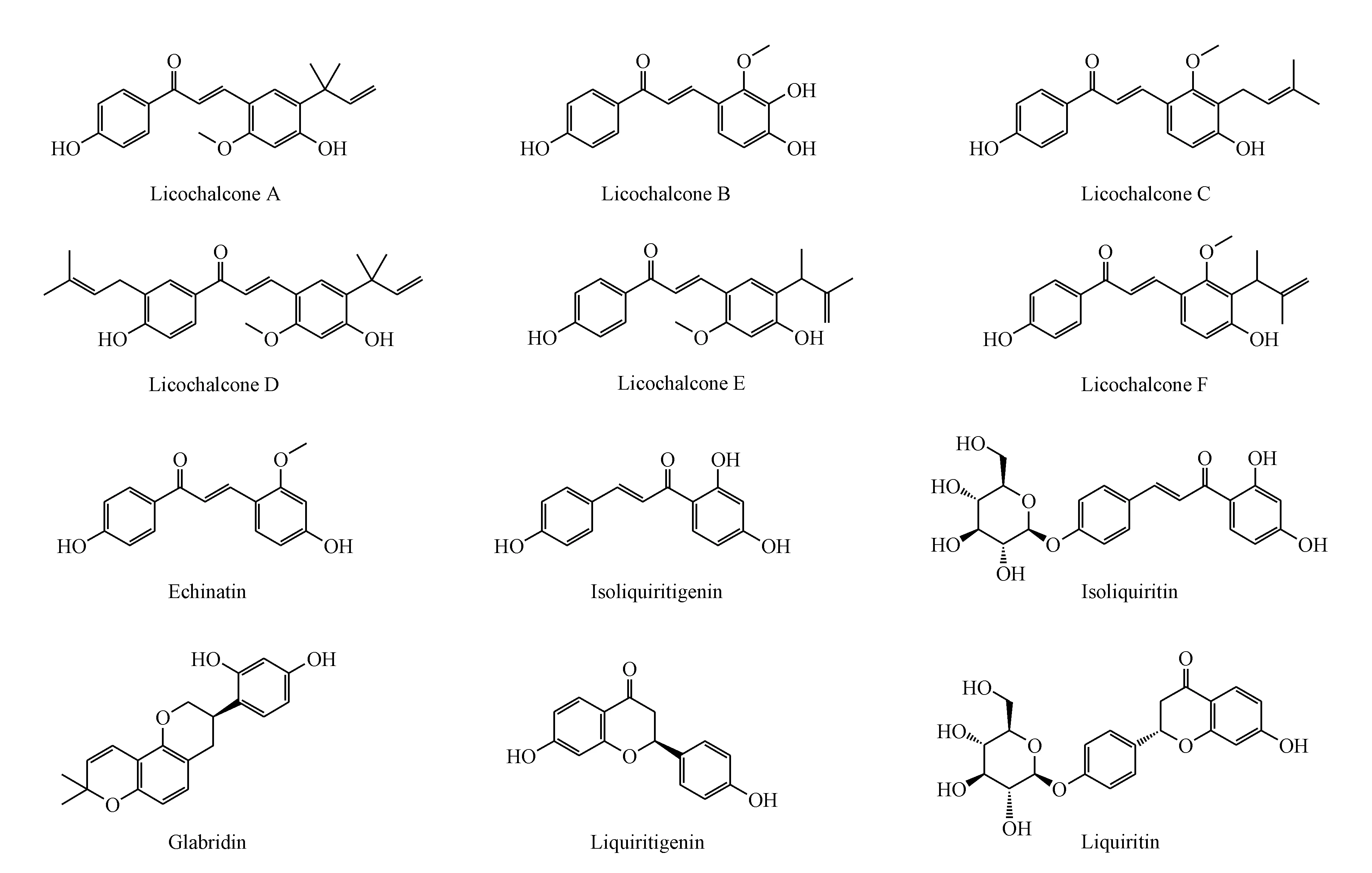
Figure1 Structures of licorice flavonoids
1 材 料
1.1 试 剂
甘草查耳酮A-F、刺甘草查耳酮、异甘草苷购自北京伊诺凯科技有限公司(质量分数≥95%);甘草素、异甘草素、甘草苷购自百灵威科技有限公司(质量分数≥95%);光甘草定、夫拉平度(flavopiridol)购自阿拉丁试剂有限公司(质量分数≥95%);Kinase-Glo®Luminescent Kinase Assay试剂盒购自上海浩然生物技术有限公司;CCK-8试剂盒购自武汉博士德生物工程有限公司;CDK1/cyclin B购自英国SignalChem公司;其他试剂购自国药集团化学试剂有限公司(质量分数≥95%);DMEM、MEM培养基、胎牛血清购自美国Hyclone公司。
1.2 仪 器
ELx808吸收光酶标仪(美国BioTek公司);C6流式细胞仪(美国BD公司);FA1004B分析天平(上海越平科学仪器有限公司);数显游标卡尺(美国耐美特公司公司)。
1.3 动物与细胞
BALB/c裸小鼠,体重14~17 g,雌雄各半,由武汉大学动物实验中心提供,动物使用许可证:SCXK(鄂)2008-0004;动物级别:SPF级。细胞株:人正常肝细胞HHL-5、人肝癌细胞Bel-7402细胞购自武汉大学细胞典藏中心;人正常肝细胞LO2购自中国协和医科大学基础医学研究所基础医学细胞中心;非洲绿猴肾细胞Vero购自中国科学院典型培养物保藏委员会细胞库。
2 方法与结果
2.1 CDK1活性测试
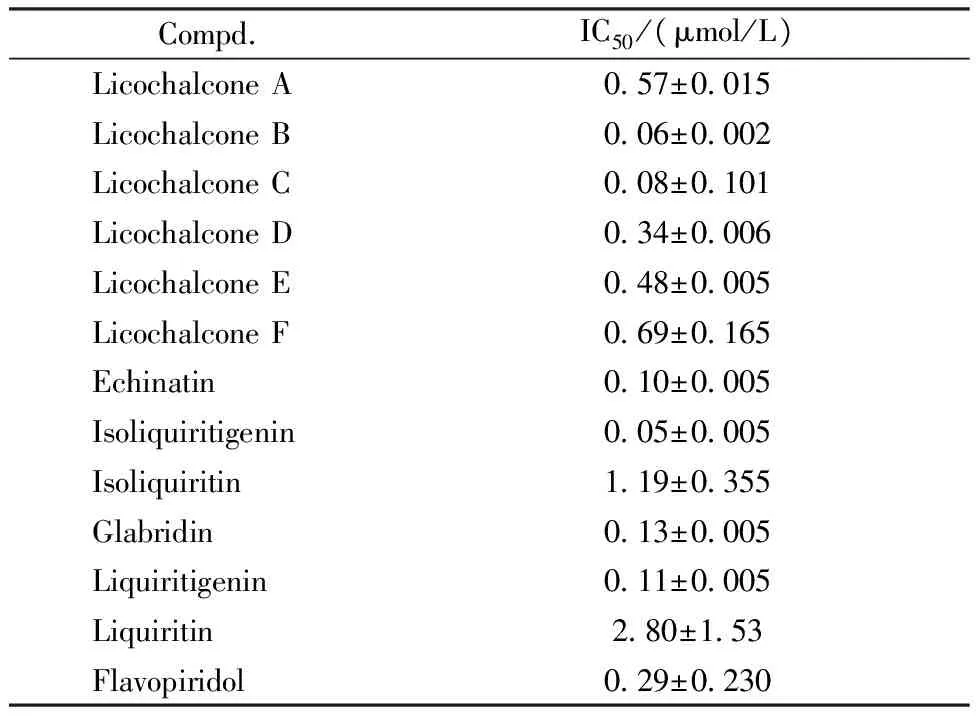
使用Kinase-Glo®Luminescent Kinase Assay试剂盒测试甘草黄酮类化合物对CDK的抑制活性,以夫拉平度为阳性对照,按照操作说明书在测试板中每孔加入激酶CDK1/cyclinB、ATP-荧光底物和不同浓度的待测物,将测试板在30 ℃下反应1 h后,每孔再加入Kinasw Glo Plus并继续在30 ℃下放置20 min。使用酶标仪读取发光强度,分析实验结果,并计算出IC50,以此判断甘草黄酮类化合物对细胞周期依赖性蛋白激酶的抑制活性,结果见表1。

Compd.IC50/(μmol/L)LicochalconeA057±0015LicochalconeB006±0002LicochalconeC008±0101LicochalconeD034±0006LicochalconeE048±0005LicochalconeF069±0165Echinatin010±0005Isoliquiritigenin005±0005Isoliquiritin119±0355Glabridin013±0005Liquiritigenin011±0005Liquiritin280±153Flavopiridol029±0230
总的来说,这些甘草黄酮化合物对CDK1/cyclin B均有较强的抑制活性。但是,两个黄酮苷化合物(异甘草苷和甘草苷)的活性低于其他黄酮类化合物。事实上,异甘草苷和甘草苷抑制CDK1/cyclin B的IC50均在微摩尔水平,而其他甘草黄酮抑制CDK1/cyclin B的IC50均在纳摩尔水平。尤其是异甘草素(IC50=0.05±0.005 μmol/L)对CDK1/cyclin B的抑制活性是阳性药物夫拉平度(IC50=0.29±0.230 μmol/L)的近6倍。
2.2 分子对接
CDK1的晶体结构目前还未解析出来,按照参考文献以CDK2晶体结构(PDB code:1YKR)为模板对CDK1进行同源建模[9-11]。通过ChemBio 3D ultra 14.0和Autodock 4.2D中的Ligand模块进行小分子结构的预处理。对接软件使用Autodock4.2D,利用晶体结构中的配体定义口袋盒子,对接盒子边长设置为30 Å,使用半经验自由能进行评价,拉马克遗传算法循环100次,其余参数保持默认,结果见图2。
在CDK1的活性口袋中,阳性药物夫拉平度能够与氨基酸残基E81、L82、S84、Q132和D149相互作用形成5个氢键(图2-A),其中夫拉平度与E81、L82和D149的氢键作用是抑制CDK1的关键作用力[10];而异甘草素在CDK1的活性口袋中也能够与氨基酸残基E81、L82和D149形成氢键(图2-B),因此异甘草素对CDK1也有较强的抑制活性。此外,异甘草素还能与K33、S84和D86相互作用形成氢键,总共与CDK1的活性口袋氨基酸残基形成了6个氢键。显然,异甘草素在CDK1的活性口袋较夫拉平度与更多的氨基酸相互作用,因此异甘草素对CDK1的抑制活性强于阳性药物夫拉平度。
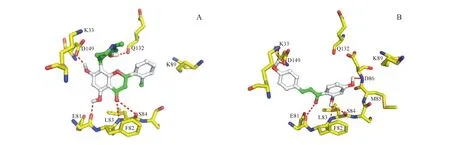
Figure2 Predicted binding modes of flavopiridol-CDK1 and isoliquiritigenin-CDK1
A:Docking poses of flavopiridol-CDK1,which can form hydrogen bonds with residues E81,L83,S84,Q132,D149;B:Docking poses of isoliquiritigenin-CDK1,which can form hydrogen bonds with residues K33,E81,L83,S84,D86,D149
2.3 体外抗增殖活性
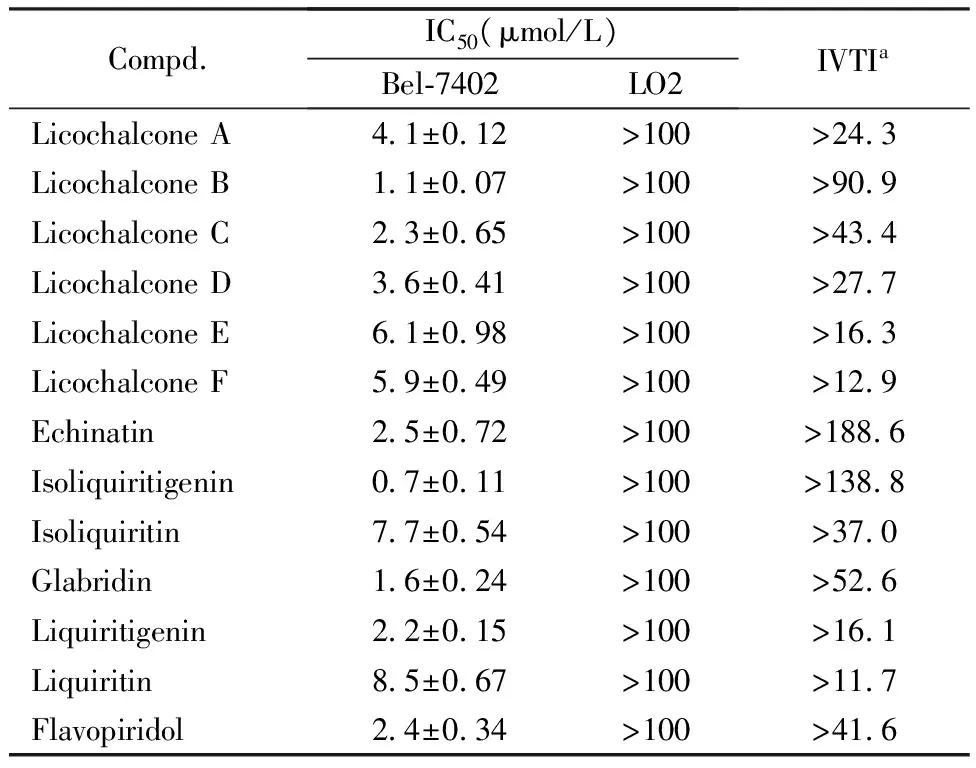
选取人肝癌细胞Bel-7402为测试细胞株,正常的肝细胞LO2、HHL-5以及非洲绿猴肾细胞Vero为对照细胞。以CDK1抑制剂夫拉平度为阳性对照药,采用CCK-8试剂盒对甘草黄酮进行抗肿瘤活性评价。取对数生长期的测试细胞株悬浮于含10%胎牛血清的培养基中,铺至96孔培养板中。待细胞完全贴壁后,弃去原培养液,加入含有测试药物的培养液100 μL培养48 h,弃去原培养液,每孔加入CCK-8溶液10 μL,在培养箱中继续孵育4 h,用酶标仪在490 nm测定每孔的吸收度,分析实验结果,并计算出IC50,结果见表2。

Compd.IC50(μmol/L)Bel⁃7402LO2IVTIaLicochalconeA41±012>100>243LicochalconeB11±007>100>909LicochalconeC23±065>100>434LicochalconeD36±041>100>277LicochalconeE61±098>100>163LicochalconeF59±049>100>129Echinatin25±072>100>1886Isoliquiritigenin07±011>100>1388Isoliquiritin77±054>100>370Glabridin16±024>100>526Liquiritigenin22±015>100>161Liquiritin85±067>100>117Flavopiridol24±034>100>416
aIVTI:invitrotherapeutic index,ratio of IC50(LO2) to IC50(Bel-7402)
结果(表2)表明,12个甘草黄酮化合物对肝癌Bel-7402细胞均有较强的抑制作用,其IC50均小于10.0 mol/L,其中甘草查耳酮B、异甘草素和光甘草定对Bel-7402的抑制活性强于阳性药物夫拉平度,特别是异甘草素抑制Bel-7402的IC50达到了纳摩尔水平,是阳性药物夫拉平度的3倍以上。
为了进一步评价甘草黄酮化合物的毒性,用正常的肝细胞LO2作为测试细胞株来检测其毒性,并计算出选择性指数(invitrotherapeutic index,IVTI),化合物的IVTI越高,安全性越高[12]。从表2中可以发现,这类甘草黄酮基本没有毒性(IC50>100 mol/L),并有较高的IVTI,其中异甘草素的治疗指数(IVTI>138)显著高于其他甘草黄酮化合物和阳性药物夫拉平度。但是,异甘草素对另外一种正常的肝细胞HHL-5(IC50=6.0±0.18 μmol/L)和非洲绿猴肾细胞Vero(IC50=3.6±0.58 μmol/L)均有较强的毒性作用(图3)。
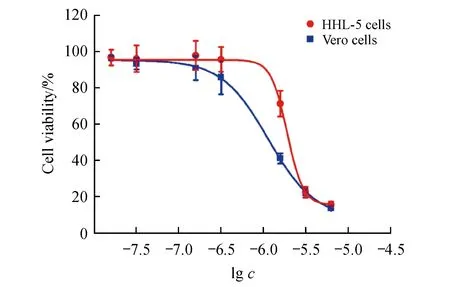
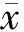
2.4 异甘草素对Bel-7402细胞凋亡的影响
取对数生长期的Bel-7402细胞株悬浮于含10%胎牛血清的DMEM培养基中,铺至12孔培养板中,置37 ℃、5% CO2培养箱中培养。分为测试组、阳性药物对照组和空白对照组。待细胞完全贴壁后,弃去原培养液,测试组和阳性药物分别加入含0.7 μmol/L异甘草素和夫拉平度的培养液1 mL,继续培养24 h。按照FITC Annexin V/7-AAD试剂盒的说明书收集细胞,冰的PBS洗涤3次,离心弃去上清液,加入1×结合缓冲液100 μL,分别加入FITC Annexin V 5 μL和7-AAD 5 μL,混匀后室温避光15 min,上流式细胞检测仪进行检测,分析Bel-7402细胞凋亡率,结果见图4。
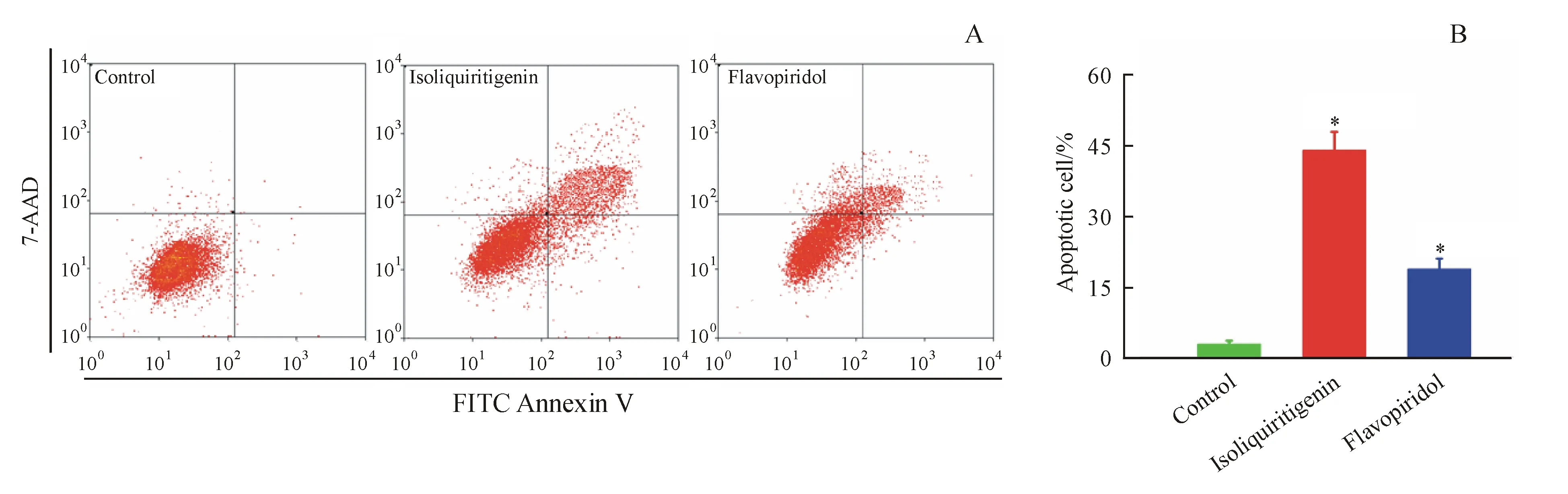
Figure4 Isoliquiritigenin induce Bel-7402 cell apoptosisinvitro

图4-A中右下象限和右上象限分别代表早期凋亡和晚期凋亡,而将两象限的百分率相加即为发生凋亡的细胞比率。结果显示,与空白对照组相比,异甘草素和夫拉平度均能显著地诱导Bel-7402细胞凋亡,但异甘草素诱导Bel-7402细胞的凋亡作用强于夫拉平度,其凋亡率分别为44.1%和19.2%(图4-B)。
2.5 急性毒性实验
在表2中,异甘草素对正常的肝细胞HHL-5和非洲绿猴肾细胞Vero有毒性。因此,为了进一步探讨异甘草素在动物体内是否有毒性,按照参考文献的方法[13],选取4~5周龄的裸鼠48只,随机将其分为6组不同的给药剂量组,给药剂量分别为2.17,2.80,3.61,4.65,5.99,7.72 mg/kg,每组8只,雌雄各半,在灌胃给药后1 h、5 h、3 d、4~18 d观察记录实验小鼠的存活状态,并用寇氏法计算出异甘草素的LD50及LD50的95%可信限,结果如表3所示。
Table3 Acute toxicity of isoliquiritigenin in mice
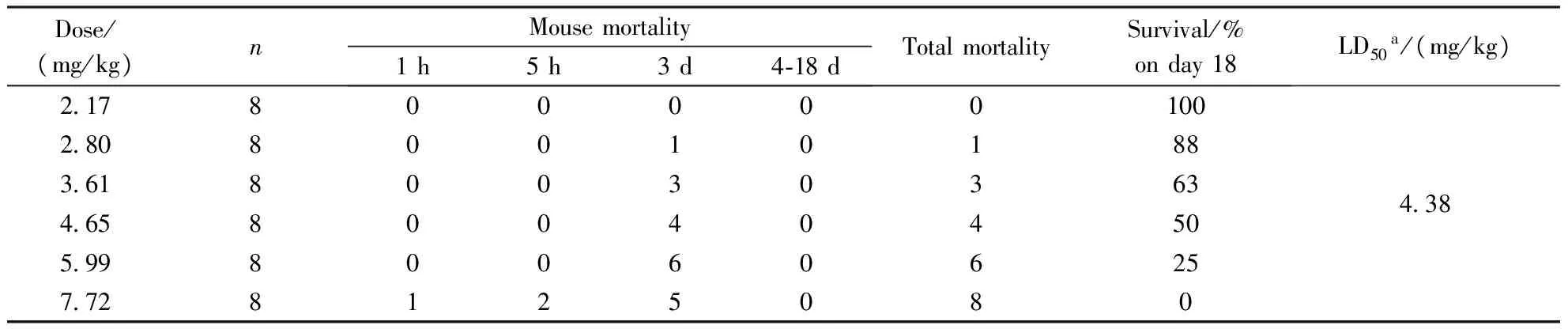
Dose/(mg/kg)nMousemortality1h5h3d4⁃18dTotalmortalitySurvival/%onday18LD50a/(mg/kg)217800000100280800101883618003036346580040450599800606257728125080438
a95% Confidence limits:3.78-5.07 mg/kg
当给予小鼠2.17 mg/kg剂量时,小鼠在18 d内存活率为100%;而给予大剂量7.72 mg/kg剂量时,3 d内全部死亡。根据不同剂量组的小鼠死亡率,计算出了异甘草素的LD50为4.38 mg/kg,其95%的可信限为3.78~5.07 mg/kg。
2.6 体内抗肿瘤活性
从表2和图3的体外抗肿瘤活性实验结果可以看出,异甘草素对肝癌Bel-7402细胞展现出了较强的抑制活性。因此,本研究建立了肝癌Bel-7402裸鼠皮下瘤模型,来进一步考察异甘草素的动物体内抗肝癌活性。选取4~5周龄的裸鼠在其右侧肩部皮下接种Bel-7402的细胞混悬液,每只100 μL(4×106~5×106个细胞),接种1周后,随机对小鼠分为治疗组、阳性药物组和空白对照组,每组4只,雌雄各半,灌胃给药。通过急性毒性实验本研究发现异甘草素经口给药剂量为2.80 mg/kg在第3天将引起实验裸鼠死亡,而在剂量为2.17 mg/kg不会造成实验裸鼠死亡。因此,为了保证在给药治疗期间实验裸鼠的存活率,治疗组异甘草素和阳性药物组夫拉平度的给药剂量均为2.17 mg/(kg·d),其中空白对照组给予等体积的PBS,连续给药18 d。每隔1天称量裸鼠的体重,并测试肿瘤长宽,计算肿瘤体积。治疗结束后将裸鼠处死,取下肿瘤并称重,结果见图5。
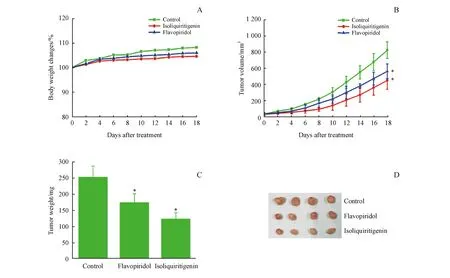
A:body weight;B:tumor volume;C:tumor weight;D:tumor size
*P<0.01vscontrol group
异甘草素和阳性药物夫拉平度[2.17 mg/(kg·d)]经灌胃连续给药18 d,给药期间实验裸鼠的体重在逐渐地增加(图5-A)。从肿瘤体积随时间变化曲线(图5-B)可以发现,与对照组相比,异甘草素和阳性药物夫拉平度在裸鼠体内均能够有效地减慢肿瘤的增长,其中异甘草素的效果更佳。实验结束后,取下的实验小鼠肿瘤也是异甘草素给药组小鼠的肿瘤重量(图5-C)和肿瘤体积均小于阳性药物夫拉平度组(图5-D)。
3 讨 论
本研究结果显示,这些甘草黄酮对CDK1具有靶向性。但是,甘草黄酮化合物之间对酶的抑制活性却有较大的差异。从结构上看,两个甘草黄酮苷(异甘草苷和甘草苷)对CDK1的抑制活性低于其相应的甘草黄酮,这说明甘草黄酮上与糖基相连的羟基是药效团。从分子对接的结果看,与糖基相连的羟基能够与酶形成氢键作用,而形成糖苷后不能形成氢键,因此甘草黄酮苷的CDK1酶抑制活性低于甘草黄酮。在甘草查耳酮A-F、刺甘草查耳酮、异甘草素、光甘草定、甘草素中,甘草查耳酮A-F、刺甘草查耳酮、异甘草素属于黄酮的另一个分支——查耳酮,这类8个化合物具有α,β-不饱和酮结构,有较好的柔性,能够与多种受体结合,也是一个抗肿瘤的药效团,因此甘草查耳酮A-F、刺甘草查耳酮、异甘草素的酶抑制活性要强于光甘草定和甘草素。但是,其α,β-不饱和酮的位置有变化,其中异甘草素的α,β-不饱和酮的羰基与间苯二酚相连,而该羰基能够同间苯二酚的羟基一起与酶的氨基酸残基形成氢键,因此异甘草素的酶抑制活性强于其他类型甘草黄酮。
在体外抗肿瘤活性中,这类甘草黄酮对肝癌Bel-7402有较强的抑制活性,其抑制Bel-7402的趋势与酶抑制活性一致,也是甘草黄酮(甘草查耳酮A-F、刺甘草查耳酮、异甘草素、光甘草定、甘草素)强于黄酮苷(异甘草苷和甘草苷),具有α,β-不饱和酮的化合物甘草查耳酮A-F、刺甘草查耳酮、异甘草素强于化合物光甘草定、甘草素。其中异甘草素(IC50=0.7±0.11 mol/L)对Bel-7402展现出了最强的抑制活性,是阳性药物夫拉平度(IC50=2.4±0.34 mol/L)的3倍。
尽管这类甘草黄酮对正常的肝LO2细胞没有毒性,但是异甘草素对另一种正常的肝细胞HHL-5(IC50=6±0.18 μmol/L)和非洲绿猴肾细胞Vero(IC50=3.6±0.58 μmol/L)均有较强的毒性作用(图3)。进一步研究发现异甘草素对小鼠的LD50为4.38 mg/kg。异甘草素对LO2细胞没有毒性,可能是由于LO2细胞系是在人正常肝细胞原代培养的基础上经过特殊处理而获得的细胞,该细胞株已经永生化,并不是正常的肝细胞,也不是原代细胞,只是其生长和死亡的规律和正常肝细胞较为接近,但已不具备很多正常细胞的特性,因此异甘草素对LO2没有毒性,而采用正常肝细胞HHL-5更能反映出异甘草素内在毒性,实验结果(表3)表明异甘草素对HHL-5细胞有较强的毒性,在动物体内也显示毒性较大。在动物体内实验中,异甘草素也能够有效地抑制肿瘤的增长,其抑瘤率为51.0%,是阳性药物夫拉平度(31.0%)的1.6倍。
总之,甘草黄酮是一类细胞周期依赖性蛋白激酶(CDKs)抑制剂,其中异甘草素可以作为先导物进行结构修饰和改造,以获得抗肝癌活性高、毒性较小的化合物,值得进一步深入研究。
[1] He W,Ning J,Wu JJ,etal.Research progress in interaction between chemical components of Glycyrrhizae Radix and cytochrome P450 enzyme[J].ChinTraditHerbDrugs(中草药),2016,47(11):1974-1981.
[2] Han YD,Wang B,Wang ZY,etal.Recent research progress in pharmacological effects of glycyrrhizic acid[J].ChinNewDrugsJ(中国新药杂志),2012,21(21):2499-2505.
[3] Jin SX,Jin SY,Li XY,etal.Preparation and in vivo evaluation of glycgrrhizin acid-sodium deoxycholate/phospholipid-mixed mi-celles[J].ChinPharmJ(中国药学杂志),2013,48(4):280-285.
[4] Cui YR,Li DF,Ju B,etal.Antitumor effects of four licoflavonesinvitro[J].FoodSciTechnol(食品科技),2010,35(7):88-92.
[5] Mourboul A,Wang YB,Xu FY,etal.Anticancer activity of flavonoids from Xinjiang glycyrrhiza glabra in human Bel-7402 hepatocarcinoma cell line[J].JXinjiangMedUniv(新疆医科大学报),2013,36(12):1744-1748.
[6] Xu FY.Anticancer activity and mechanism of actions of glycyrrhetinic acid compounds and licorice chalcone derivatives in Human hepatocarcinoma Bel-7402 cells[D].Xinjiang:XinjiangMedUniv(新疆:新疆医科大学),2013.
[7] Huang W,Chen Q,Yang WC,etal.Efficient synthesis and antiproliferative activity of novel thioether-substituted flavonoids[J].EurJMedChem,2013,66:161-170.
[8] Tsaur I,Makarevic J,Hudak L,etal.The cdk1-cyclin B complex is involved in everolimus triggered resistance in the PC3 prostate cancer cell line[J].CancerLett,2011,313(1):84-90.
[9] Hamdouchli C,Zhou B,Mendoza J,etal.Structure-based design of a new class of highly selective aminoimidazo[1,2-a]pyridine-based inhibitors of cyclin dependent kinases[J].BioorgMedChem,2005,15(7):1943-1947.
[10] Nguyeng B,Lozach O,Wang Q,etal.Synthesis,biological evaluation,and molecular modeling of natural and unnatural flavonoidal alkaloids,inhibitors of kinases[J].JMedChem,2012,55(6):2811-2819.
[11] Zhang QQ,Yao QZ,Zhang SP,etal.Homology modeling,molecular docking,and 3D-QSAR of indirubin analogues as CDK1 inhibitors[J].ActaPhys-ChimSin(物理化学学报),2014,30(2):371-381.
[12] Muller PK,Molton MN.The determination and interpretation of the therapeutic index in drug development[J].NatRevDrugDiscov,2012,11(10):751-761.
[13] Ai Y,Hu Y,Kang FH,etal.Synthesis and biological evaluation of novel olean-28,13β-lactams as potential antiprostate cancer agents[J].JMedChem,2015,58(11):4506-4520.
