基于p38MAPK表达的烟草毒理学评价
2018-03-19魏克强杨进文
张 璇,魏克强,杨进文
(1.山西大学生命科学学院,山西太原 030006;2.山西农业大学农学院,山西太谷 030801)
烟草的化学成分直接影响卷烟制品的安全性和可用性,其形成与品种类型、栽培措施、生态环境等因素密切相关[1-3]。研究表明,不同基因型烟草品种的化学成分存在较大差异;香烟烟雾气溶胶含有近5 000种化学成分,其中,1/3直接来源于烟草本身,而2/3则是在香烟的燃烧、裂解、蒸馏、冷凝等一系列物理化学作用中新产生的[4]。采用体外毒理学评价方法,即哺乳动物细胞系细胞毒性分析(中性红试验)、细菌诱变分析(Ames试验)和细胞遗传学分析(微核试验),以及“特定机体损伤”即慢性阻塞性肺疾病(COPD)的动物模型评价方法,山西大学生命科学学院实验室发现不同基因型烟草的毒理学效应差异显著[5-7],这对培育低毒少害的烟草新品种具有一定的指导意义。
COPD是一种以持续性气流受限为特征的慢性呼吸系统疾病,吸烟被认为是诱发COPD最主要的危险因素[8-9],其致病机制涉及免疫应答失衡、氧化/抗氧化失衡、蛋白酶/抗蛋白酶失衡、细胞增殖/凋亡失衡等,在疾病进程中多种炎症细胞、细胞因子及转录因子、信号通路发挥了重要作用[10-11]。特别是,p38丝裂原活化蛋白激酶(p38MAPK)参与了细胞应激、炎症反应、细胞凋亡等生理和病理过程,是多条信号途径的交汇点和共同通道,其表达的变化有可能作为评价烟草毒理学效应的潜在标志物[12-13]。
本研究以2个基因型的烟草品种为材料,采用单纯烟熏法构建出大鼠的COPD模型,通过H.E.染色、组织病理学分析以及p38、磷酸化p38(p-p38)的免疫组化分析,评价了不同基因型烟草对大鼠肺组织的毒理学效应,旨在为揭示空气污染物暴露的毒理学机制、建立标准的香烟烟雾毒理学评价方法提供理论依据。
1 材料和方法
1.1 试验材料
1.1.1 试验动物 健康清洁级雄性SD大鼠24只(5周龄,体质量约185 g),购自军事医学科学院实验动物中心。试验前在恒温恒湿条件下暂养7 d,自由饮水与进食。2个基因型烟草品种,由山西农业大学烟草育种研究室提供,其烟叶由山西昆明烟草有限责任公司分别卷制成单料烟Y-1和Y-2。
1.1.2 主要试剂与仪器 兔抗大鼠p38,p-p38多克隆抗体,分别购自Proteintech Grope及爱必信(上海)生物科技有限公司;山羊抗兔IgG SABC免疫组化染色试剂盒,购自武汉博士德生物工程有限公司;浓缩型DAB试剂盒,购自北京中杉金桥生物技术有限公司;戊巴比妥钠、多聚甲醛、乙醇、二甲苯、伊红、苏木精等均为国产分析纯。
仪器主要有石蜡切片机(Leica RM2255,德国);ZKPJ-1A展烤片机(天津天利航空机电有限公司);光学显微镜(Olympus BX51,日本);HHS-21-4型电热恒温水浴锅(上海博迅实业有限公司)等。
1.2 试验方法
1.2.1 COPD模型构建及采样 将大鼠随机分为3组:健康对照组(CK)、Y-1组、Y-2组,每组 8只。CK组大鼠仅吸入新鲜空气,Y-1,Y-2组大鼠置于染毒箱(60 cm×80 cm×100 cm)内,分别以卷制的单料烟Y-1,Y-2进行60 d的烟雾暴露染毒:每组同时点燃10支烟,通过气泵和导管装置将烟雾导入染毒箱,持续暴露染毒1 h,每天2次,每次间隔4 h,每周染毒6 d。分别于染毒30,60 d后,每组随机取4只大鼠,腹腔注射1%戊巴比妥钠(3 mL/kg)麻醉,解剖取右肺组织,用预冷的生理盐水冲洗干净,置于4%多聚甲醛中固定。
1.2.2 肺组织H.E.染色及病理评分 将固定的肺组织行常规石蜡包埋、切片(5 μm)、H.E.染色。参考文献 [14]的方法,每张切片随机选取5个视野于100倍光镜下观察肺组织的病理改变,并从7个方面进行评分:(1)气道腔阻塞;(2)气道上皮糜烂脱落;(3)气道上皮杯状化生;(4)气道上皮鳞状化生;(5)气道壁炎症细胞浸润;(6)气道壁纤维结缔组织增生;(7)气道壁平滑肌增生。以上每项从无明显改变到严重改变分别用0~4分表示,各项评分合计为一个视野的病理评分。
1.2.3 免疫组化法检测 石蜡切片常规脱蜡,免疫组化三步法(SABC法)检测p38,p-p38的表达,DAB显色,苏木精复染。每张切片中随机选取5个视野,使用Image-ProPlus 6.0软件读取每个视野下阳性物质表达区域的平均光密度值。
1.3 数据处理
应用SPSS 17.0软件处理数据,所有计数资料以“均数±标准差(±s)”表示;多组间比较采用单因素方差分析(One-Way ANOVA),P<0.05表示差异具有统计学意义。
2 结果与分析
2.1 肺组织H.E.染色及病理学评分
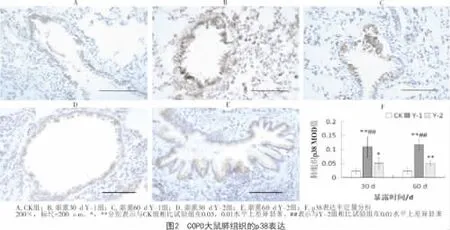
观察结果显示,CK组鲜见炎症细胞聚集,气道上皮完整,管腔内无黏液分布,管壁无增厚。随烟雾暴露时间的延长,Y-1组和Y-2组呈现出不同程度的病理改变:暴露30 d后,小气道周围出现少量聚集的炎症细胞,气道上皮轻微脱落,上皮细胞出现鳞状和杯状细胞化生,管腔内偶见黏液,气道壁结缔组织和平滑肌开始增生;烟熏60 d后,Y-1组与Y-2组气道周围出现大量聚集的炎症细胞,气道上皮脱落严重,上皮细胞化生现象频繁,细胞排列不规则,黏膜皱襞增多、黏液分泌导致气道狭窄、堵塞,气道壁明显增厚,表现出COPD典型的病理特征。病理学评分显示,与CK组相比,Y-1组与Y-2组的病理改变显著(P<0.01);与Y-2组相比,Y-1组的病理改变更为严重,但差异不显著(P>0.05)。这表明,不同基因型烟草释放的烟雾对COPD进程有不同程度的影响(图1)。
2.2 肺组织p38及p-p38的表达
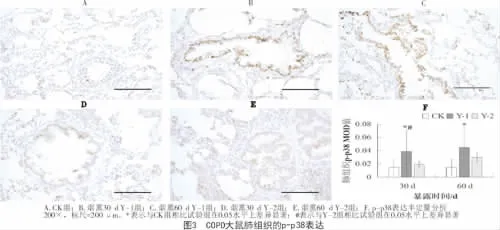
免疫组化分析表明,在60 d的疾病进程中,各处理组COPD大鼠肺组织的p38表达水平变化不显著(P>0.05);但与 CK组相比,Y-1组、Y-2组的p38表达水平均显著升高,且Y-1组明显高于Y-2组(P<0.05)。随暴露时间的延长,Y-1组、Y-2组的p38磷酸化水平逐渐升高;与CK组相比,Y-1组的p-p38表达明显上调(P<0.05);在烟熏30 d时,Y-1组p-p38的表达明显高于Y-2组(P<0.05)。表明香烟烟雾能够激活大鼠肺组织p38的表达及其磷酸化,且p38的表达与活化受不同基因型烟草释放烟雾的明显影响。

3 讨论
丝裂原活化蛋白激酶(MAPK)级联信号转导通路是哺乳动物介导细胞反应的重要信号传导系统。MAPK家族主要有3个成员,即细胞外信号调节蛋白激酶(ERK)、应激活化蛋白激酶(JUNK)和蛋白激酶p38。其中,p38是细胞内信号传导的经典途径,在应激条件下可以被激活而磷酸化,进入细胞核内或转移到其他部位,发挥对氧化应激、炎症反应以及细胞凋亡的调控[15-16]。
气道或肺组织对香烟烟雾有害成分或颗粒的异常反应是诱发COPD的最主要原因[8-9]。香烟烟雾能够损伤肺泡上皮细胞,受损的肺泡上皮细胞产生炎症介质,将树突细胞、肺泡巨噬细胞和中性粒细胞趋化到损伤部位并激活,分泌蛋白酶进一步损伤肺组织[17],这一过程可以诱导p38磷酸化,在疾病进程中发挥不可或缺的作用。而抑制p38 MAPK的激活能有效改善COPD患者的肺功能、降低炎症介质的水平,提示p38可能是药物治疗COPD的潜在靶位点[18-20]。
本研究表明,2个基因型烟草染毒的大鼠,在COPD发病进程中其肺组织的病理变化程度与p38表达及其磷酸化水平的趋势基本一致,这可能是由于活化的p38促进了炎症细胞的聚集、炎症介质的释放和氧化应激的损伤。但肺组织的p38表达、活化以及病理改变与烟草材料存在密切的相关性,这可能与不同基因型烟草的化学成分存在较大差异有关,p38可能是评价烟草导致“特定机体损伤”的一个潜在标志物,这对烟草毒理学的研究具有重要的意义。
[1]杜文,谭新良,易建华,等.用烟叶化学成分进行烟叶质量评价[J].中国烟草学报,2007,13(3):25-31.
[2]孙福山,王丽卿.烟叶成熟度及烘烤关键指标与烟叶质量关系的研究[J].中国烟草科学,2002,23(3):25-27.
[3]王彦亭.中国烟草种植区划[M].北京:科学出版社,2010.
[4]RICHTER P,PECHACEK T,SWAHN M,et al.Reducing levels of toxic chemicals in cigarette smoke:A new healthy people 2010 objective[J].Public Health Reports,2008,123(1):30-38.
[5]李佳美.环境烟雾暴露对SD大鼠主要组织的损伤效应研究[D].太原:山西大学,2016.
[6]张永健,魏克强,杨进文.不同基因型烟草的细胞毒性与致突变性分析[J].山西农业科学,2017,45(3):374-378.
[7]武娟,魏克强,任建英.不同基因型烟草体内外毒理学效应的初步分析[J].山西农业科学,2016,44(5):596-599.
[8]JOHN-SCHUSTER G,GÜNTER S,HAGER K,et al.Inflammaging increases susceptibilitytocigarette smoke-induced COPD[J].Oncotarget,2016,7(21):30068-30083.
[9] CHURG A,COSIO M,WRIGHT J L.Mechanisms of cigarette smoke-induced COPD:insights from animal models[J].American Journal of Physiology Lung Cellular&Molecular Physiology,2008,294(4):612-31.
[10]MACNEE W.Pathogenesis of chronic obstructive pulmonary disease[J].Clinics in Chest Medicine,2007,28(3):479-513.
[11]DAHESHIAM.Pathogenesis ofchronic obstructive pulmonarydisease(COPD)[J].Clinical&Applied ImmunologyReviews,2005,5(5):339-351.
[12]RENDA T,BARALDO S,PELAIA G,et al.Increased activation of p38 MAPK in COPD[J].European Respiratory Journal,2008,31(1):62.
[13]NATH P,LEUNGSY,WILLIAMSA,et al.Importance ofp38 mitogenactivated protein kinase pathway in allergic airway remodelling and bronchial hyperresponsiveness[J].European Journal of Pharmacology,2006,544(1/3):160-167.
[14] COSIO M,GHEZZO H,HOGG J C,et al.The relations between structural changes in small airways and pulmonary-function tests[J].NewEngland Journal ofMedicine,1978,298(23):1277.
[15] SUN S J,WU X P,SONG H L,et al.Baicalin ameliorates isoproterenol-induced acute myocardial infarction through iNOS,inflammation,oxidative stress and p38MAPK pathwayin rat[J].International Journal of Clinical&Experimental Medicine,2015,8(12):22063.
[16]NAN Y,CHANG R,JIANG H,et al.Downregulation of p38 phosphorylation correlates with low-grade differentiation and proliferation of lung squamous cell carcinoma[J].American Journal of Translational Research,2017,9(4):1922.
[17]赵开顺.吸烟COPD患者清道夫受体A(SR-A)介导炎症的影响[D].上海:上海交通大学,2015.
[18] GAFFEY K,REYNOLDS S,PLUMB J,et al.Increased phosphorylated p38 mitogen-activated protein kinase in COPD lungs[J].European RespiratoryJournal,2013,42(1):28-41.
[19]黄翠萍,徐旭燕.慢性阻塞性肺疾病患者p38蛋白激酶表达的变化及其临床意义[J].中国老年学,2012,32(2):233-234.
[20]吴洁,戴伟,辛晓峰.P38MAPK在慢性阻塞性肺疾病中的作用[J].临床肺科杂志,2015(1):138-141.
