聚合物分离膜材料的制备与结构调控
2018-03-17唐安琪王章慧张梦晓路景驭朱利平
唐安琪,王章慧,张梦晓,路景驭,朱利平
(浙江大学高分子科学与工程学系 高分子合成与功能构造教育部重点实验室,浙江 杭州 310027)
1 前 言
随着我国经济的飞速发展和城市化进程的不断加快,水污染和水资源缺乏问题日益加剧,加强水资源保护、不断革新水处理技术,是实现可持续发展道路上重要而紧迫的任务。膜技术作为一项新型高效的分离技术,被广泛应用于水处理、食品饮料纯化、制药工程、医疗过滤、空气净化、工业分离、锂电池/燃料电池隔膜等领域。膜材料是膜技术的核心,目前绝大多数膜技术都依赖于有机高分子膜。近年来,以提高传统分离膜材料渗透性、选择性、抗污染性能、机械强度等为特征的高性能化,正成为膜技术领域新的发展趋势。通过制膜原材料的设计和制膜方法的优化,对膜材料的多层次微结构进行调控,低成本地实现膜材料的高性能化,是膜技术进一步推广应用中亟待解决的关键问题之一[1]。本文从膜材料的制备方法与改性两方面着手,介绍了近年来聚合物膜材料制备与结构调控方面的研究进展。
2 膜材料制备方法研究进展
聚合物膜材料的主要制备方法包括相转化法(分为非溶剂致相分离法(NIPS)、热致相分离法、溶剂蒸发法、蒸气相沉淀、蒸汽诱导相分离等)、拉伸法、烧结法、径迹蚀刻法、界面聚合法、表面自组装法等[2]。其中NIPS法是目前超/微滤膜的主流制备方法,界面聚合法是纳滤/反渗透膜的主要制备方法。本节将着重介绍相转化法、界面聚合法、自组装法的一些最新研究进展,并介绍一些新的制膜方法。
2.1 相转化法
1963年,Loeb和Sourirajan[3]发明了浸没沉淀相转化制膜方法,此后被广泛研究和使用,成为膜技术发展史上的里程碑。可采用相转化法制膜的聚合物主要有聚偏氟乙烯(PVDF)、聚砜(PSF)、聚醚砜(PES)、聚氯乙烯(PVC)、聚丙烯腈(PAN)、醋酸纤维素(CA)、聚乳酸(PLA)等,相转化法既可以制备小孔或无孔的致密膜,用于纳滤、反渗透和气体分离等过程,也可以制备孔径较大的超滤膜和微滤膜。
相转化法是一种相对成熟的制膜方法,近年来关于相转化法制膜的研究主要集中在采用一些新型的成膜聚合物或凝胶剂进行膜结构的调控方面。采用超临界流体成膜是一项很有意义的工作[4],该方法使用超临界流体(通常是CO2)作为非溶剂渗入聚合物内部,引发分相成孔。由于超临界CO2能够降低几乎所有的有机溶剂的内聚能,有机溶剂在超临界CO2中的扩散系数比传统的液体溶剂高1~2 个数量级,因此有利于得到具有纳米级孔道的分离膜材料。而且超临界CO2在使聚合物溶液发生相分离的同时,会对所形成的微孔膜起到干燥作用,不产生气-液相界面,从而省略了后续的干燥工艺,避免了膜干燥过程中容易引起的结构坍塌问题;此外还具有传质速率快、环境友好、便于回收等优势。超临界CO2的使用方法主要包括作为添加剂加入膜材料的制备过程、直接作为介质进行聚合反应、作为溶剂溶胀聚合物得到混合或复合膜材料等。CO2对聚合物增塑作用的大小和在本体材料中向外扩散的速率是最终影响膜孔结构形态的两个基本因素[5]。
嵌段聚合物可在一定条件下通过相分离形成纳微米级别的长程有序结构,其易于精确调控的形貌尺寸使其在微型印刷、药物传递和能源等领域得到应用,继而延伸至分离膜材料领域。嵌段聚合物制备膜材料的优势在于有序结构带来的窄孔径分布和高孔隙率,以及通过调整嵌段结构和比例即可灵活操纵的物理化学性质,这些优点赋予了嵌段共聚物膜优良的选择性、渗透性、机械强度和力学性能[6]。1999年,Liu等[7]首次将嵌段共聚物用于薄膜材料的制备,2006年,Phillip等提出如能利用嵌段共聚物制备超滤膜材料,其选择透过性有望超越传统方法制备出的水分离膜[8],并在2009年成功实践了这一设想,使用聚乳酸-降冰片烯基乙基苯乙烯嵌段共聚物(PLA-b-P(N-s-S))在玻璃板上刮涂成膜,相分离后再刻蚀聚乳酸相的方法,制备出空腔体积高达40%、孔径14 nm、具有高规整度和高选择渗透性的超滤膜[9],并利用(PS-b-PLA)体系揭示了热力学和动力学主导下均孔膜结构的形成机理[10]。2007年,Peinemann和Abetz等[11]道了利用自组装和非溶剂诱导相分离相结合的手段(SNIPS)制备长程有序孔结构的嵌段共聚物薄膜方法。最近,本研究小组采用SNIPS法首次成功制备出结构对称的PS-b-P4VP嵌段聚合物自组装自支撑多孔膜,这种多孔膜完全由嵌段共聚物棒状胶束堆积而成,含有相互贯通的孔道,并具有pH响应特征,可通过pH的调节实现纳米粒子的分级(如图1)[12]。

图1 对称结构的pH响应型PS-b-P4VP嵌段聚合物自组装多孔膜用于纳米粒子分级[12]Fig.1 Symmetrical self-assembled PS-b-P4VP porous membranes for nanoparticles fractionalization[12]
2.2 界面聚合法
界面聚合法是目前工业上制备复合纳滤膜和反渗透膜的主要方法,它是以Morgande 相界面聚合原理为基础发展起来的。该方法利用两种反应活性很高的单体或预聚物在两个互不相容的溶剂界面处相对扩散,发生快速聚合反应(通常为缩聚),从而在支撑膜的表面形成一层薄的致密的选择层,这种复合膜被称为薄层复合膜(Thin-Film Composite, TFC)。这种方法制备的分离层具有高度交联结构,且表面荷电性高、操作简单、成本低廉,然而,由于单体反应活性过高而存在难以调控,影响因素过多,稳定性不足等缺点[13]。
目前,以界面聚合反应为方法的制膜工作重点主要集中在研究开发具有特殊选择分离能力及稳定性的复合膜。包括改变有机相单体、溶液或调整水相单体的种类和配方,以及对已有工艺进行深入细致的理论研究,优化条件和工艺,并对获得的复合膜进行后处理等。帝国理工大学Livingston等[14]通过界面聚合法制备以自具微孔聚合物为活性分离层的TFC膜,耐溶剂纳滤和气体分离,丰富了界面聚合的单体选择,为聚合层稳定性的提升和分离尺寸的精细化提供了新思路。
2.3 自组装法
分子自组装的原理是利用分子与分子或分子中的某一片段与另一片段之间的分子识别,通过非共价键相互作用形成具有特定排列顺序的分子聚合体。分子之间的非共价弱相互作用如氢键、静电力、范德华力、疏水相互作用等是发生自组装的关键。1980年,Sagiv[15]第一次利用自组装的方法制备了单分子膜。目前应用于高分子分离膜制备的主要是聚电解质层层自组装技术(Layer-By-Layer Assembly),它由德国Mainz大学的Decher[16]在1991年首先提出,近年来得到了极大的关注[17,18]。该方法是将带有相反电荷的聚阴离子和聚阳离子交替沉积在基膜上制备分离层,包括静态交替沉积和压力驱动自组装两种途径。LBL法一开始仅用于制备渗透汽化膜的研究,自2003年起开始在纳滤等其它领域得到应用,拓展了其应用和方法学。其优势在于改性层的厚度、荷电性和膜表面的粗糙度可以通过控制涂覆层数方便地调控[19],但也不可避免组装过程步骤繁琐,界面稳定性不足,制备时间长以及对基膜材料要求较苛刻等缺点。
2.4 新的制膜方法
目前所应用的小孔径分离膜多为非对称的复合膜,包含致密的皮层以提供筛分孔道,孔径较大的基底提供机械支撑。近年来,表面沉积方法在非对称复合膜的制备上受到研究者们的强烈关注。这种方法通常是在多孔基膜表面沉积具有筛分能力的分离层,分离层材料的选择以及分离层与支撑层直接的界面相互作用成为这类膜材料的制备关键。近年来发展的二维材料如氧化石墨烯(GO)等为复合膜的制备提供了新的材料选择[20]。2014年,英国学者Giem课题组[21]利用真空抽滤法制备出孔径0.2 μm的氧化石墨烯(GO)自支撑膜,由于层间密堆带来的极低摩擦系数,其对水分子相较于其他溶剂和蒸汽表现出极高的选择透过性,透水速率是氦气透过速率的1010倍。研究人员还尝试在GO层与层之间引入高聚物、纳米粒子、电解质等连接物(Cross-Linker),通过其与GO 上含氧官能团发生反应生成共价键或离子键,来达到调控层间距、改善膜在润湿条件下过滤性能的目的。已有研究表明,通过引入连接物制备的GO 膜能有效去除有机染料和金属盐离子[22,23]。除此之外,共价有机框架(COF)等二维材料也逐渐由气体分离进军水分离领域,引发了不小的关注热度。印度的Banerjee团队[24,25]通过引入模板分子诱导COF单体的有序多层次组装,利用沉积法和界面反应法制备出用于精确分子分离的自支撑膜。在近年来的研究工作中,本研究小组在贻贝足丝牢固粘附特性的仿生学思路启发下,首次报道了采用多巴胺表面沉积的方法制备复合纳滤膜。徐志康教授课题组[26]和高从堦院士课题组[27]采用多巴胺/聚乙烯亚胺共沉积体系,建立了基于“可控”沉积的复合纳滤膜分离皮层构建策略,实现了复合纳滤膜的创新制备及其多功能化。
随着3D打印技术的发展和精度的不断提高,将其应用在膜材料制备中,实现孔道尺寸均匀化、精确化和简单快速的大面积制备便成为可能。Wessling等[28,29]首先使用聚二甲基硅氧烷(PDMS)进行了这方面的尝试,并开展了一系列工作。目前3D打印的精度可控制在100 nm以下,制备规模可达到1 m2以上,打印速度可达10 mm/s[30]。
在本研究小组近年来的研究工作中,提出了“凝胶复合分离膜”的设计新理念,制备了一类以PSF多孔膜为支撑骨架,功能性凝胶为表面分离层或膜孔覆盖层的“凝胶复合分离膜”[31]。研究发现,聚合物凝胶不仅覆盖在膜表面,而且填充到膜表层孔的内部,形成选择性分离层,膜的表观平均孔径从~10 nm减小至1~3 nm,实现了从超滤膜到纳滤膜的转变,可用于高价离子脱除和有机中分子拆分,阐明了凝胶复合纳滤膜尺寸筛分、静电排斥和吸附的三重分离机制(图2)[32]。
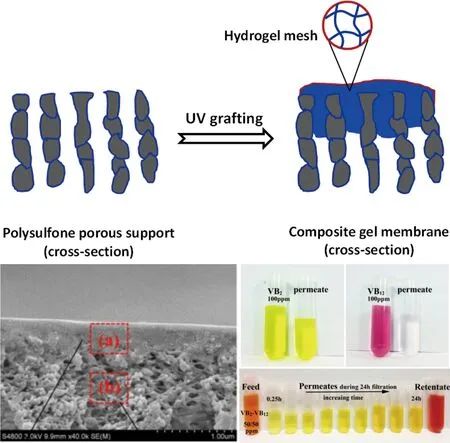
图2 UV引发表面接枝法制备具有分子分离能力的凝胶填充复合纳滤膜[32]Fig.2 Preparation of gel-filled composite nanofiltration membranes used for molecular separation[32]
3 聚合物膜材料的复合改性
膜材料的设计与制备不仅要考虑膜的渗透性、选择性、抗污染性能等特征,还要考虑材料的可加工性、机械强度、化学/热稳定性、制膜成本、环境友好程度等因素。单一材质的膜材料常常难以满足分离膜的多方面性能要求,需要整合不同材质的优势。因此,膜材料的复合改性被认为是比寻找新膜材料更加现实的途径。高分子膜材料的复合改性方法通常可以分为表面改性与本体改性两大类。
3.1 表面改性
表面改性的方法主要分为表面接枝和表面涂覆。接枝法主要是通过表面化学反应将聚合物接枝在基膜上,根据接枝方式的不同,可分为化学接枝法、等离子体接枝法和光接枝法等。通过共价键形成的改性层较为牢固,但也存在过程复杂,引入接枝基团导致膜表面孔隙率下降、粗糙度上升等问题。表面涂覆法是用沉积等手段将改性分子吸附到膜表面,形成功能化涂层。其具有操作简易,条件温和,价格经济等优点,但不足之处在于改性涂层与膜表面的相互作用力较弱,复合膜稳定性不佳,因此,近来一些研究人员尝试将改性涂层进行交联,以增加其稳定性[33]。
近年来,受贻贝足丝蛋白(Mfp)超强粘附特性启发的表面化学受到众多学者的关注,成为仿生学、化学、材料学、生物医学等领域的交叉研究热点。2007年, Messersmith研究小组[34]发现,多巴胺同时含有DOPA的邻苯二酚基团和赖氨酸的胺基官能团,在弱碱性水溶液条件下可发生氧化-自聚和交联反应,能够很好地模拟Mfp的强粘附特性,发展了一种普适的材料表面改性方法。本研究小组首先提出了多巴胺在膜材料表面自聚沉积的机理模型(图3)[35],利用聚多巴胺构建改性涂层及在此基础上接枝生物分子[36]或引发ATRP聚合[37]构建二次修饰涂层等,近年来持续开展基于贻贝仿生化学的分离功能材料制备与结构调控的研究工作,取得了一系列进展[38]。姜忠义课题组利用沉积法制备PDA/PSF复合膜,应用于渗透汽化[39]和纳滤等领域[40],同时将多巴胺分子作为水相单体参与界面聚合反应,拓展了多巴胺改性膜材料的应用形式[41]。

图3 贻贝仿生多巴胺表面沉积方法与过程示意图[32,33]Fig.3 Mussel-bioinspired dopamine surface chemistry[32,33]
3.2 本体改性
共混改性工艺改性和成膜过程一体化,具有条件温和、工艺简单、效果持久等诸多优点,但同时也不可避免地存在相容性问题,破坏有机膜的强度、韧性、抗老化性等物理和化学性质。目前常用的改性剂包括亲水性聚合物、两亲性聚合物、纳米粒子等。
亲水性聚合物如聚乙烯吡咯烷酮(PVP)、聚乙二醇(PEG)、聚甲基丙烯酸甲酯(PMMA)、聚丙烯腈(PAN)、醋酸纤维素(CA)、聚乙烯醇(PVA)等,由于其链段带有少量含氧官能团而具有部分亲水性,同时也能与疏水性膜基体材料具有较好的相容性,因此被用作传统的共混改性剂[42]。然而此类改性剂容易流失和引起本体材料的性能下降,难以取得改性效果和多相组分相容性的统一。近年来,两亲性聚合物被应用于疏水性膜材料的共混改性。其分子中同时含有疏水链和亲水链,一方面确保了与本体材料的良好相容性,另一方面赋予共混膜以更高的亲水性。研究者们发现,由于这些两亲性聚合物中亲水链与膜本体材料热力学不相容,在成膜过程中向膜-凝固浴界面迁移富集,因而显著提高了膜的表面亲水性[43,44]。但两亲性共聚物合成工艺复杂、合成产物产率过低也限制了其进一步推广使用。本课题组自2007年起开展了一系列利用两亲聚合物共混改性法调控膜结构和性能的工作,制备了具有高通量高截留率和优异抗污染性的超微滤膜材料[45];并在此工作的基础上将共混与表面反应相结合,进一步改善了膜材料的亲水性和抗污染性能[46]。姜忠义等在PVDF相转化制膜过程中同时引入两亲性聚合物和低表面能的分子刷,构建出具有梯度结构的超强抗污染和自清洁膜表面[47]。
纳米粒子独特的微观尺度效应、高比表面积和良好的润湿性一直吸引着膜改性专家和学者的目光,目前可以与有机膜材料共混并研究比较多的有新型碳纳米材料(如GO、碳纳米管等)、半导体材料(TiO2、ZnO)、Al2O3、SiO2等。近年来,纳米纤维和二维材料等新型纳米材料正在成为高效分离膜的填充改性剂,为纳米材料在分离膜中的应用打开了更广阔的视野[48]。
4 结 语
新材料与新方法的不断涌现为水处理分离膜领域的发展带来了希望与生机。机遇与挑战并存,在更深入地研究物化机理的基础上建立普适性的构效关系,通过结构设计和工艺优化实现更高的可设计性,是提高渗透性和选择性,打破二者相互制约的Trade-off难题的关键方法。进一步提高抗污染性、稳定性和溶剂条件耐受性是膜材料面向水分离实际应用的永恒主题;高性能化与功能化的深入研究是丰富膜材料应用形式、提高处理效率的重要手段。
References
[1] Xu Youyi(徐又一),Xu Zhikang(徐志康).PolymericMembraneMaterials(高分子膜材料)[M]. Beijing: Chemical Industry Press,2005:14-17.
[2] Mulder M.BasicPrinciplesofMembraneTechnology[M]. Dordrecht: Kluwer Academic Publishers, 1996: 72-75.
[3] Loeb G S,Sourirajian S.AdvancesinChemistrySeries[J], 1963,38: 117-132.
[4] Yang Feng(杨 峰),Hu Xiaoling(胡小玲),Zhao Yamei(赵亚梅),etal.ChemicalIndustryandEngineeringProgress(化工进展)[J],2007,26(2):164-167.
[5] Matsuyama H,Yamamoto A,Yano H,etal.JournalofMembraneScience[J],2002,204(1):81-87.
[6] Jackson E A,Hillmyer M A.ACSNano[J],2010, 4: 3548-3553.
[7] Liu G,Ding J,Hashimoto T,etal.ChemistryofMaterials[J], 1999, 11:2233-2240.
[8] Phillip W A,Rzayev J,Hillmyer M A,etal.JournalofMembraneScience[J],2006, 286:144-152.
[9] Phillip W A,Amendt M,O’Neill B,etal.ACSAppliedMaterialsandInterfaces[J],2009, 1:472-480.
[10] Phillip W A,Hillmyer M A,Cussler E.Macromolecules[J], 2010, 43:7763-7770.
[11] Peinemann K V,Abetz V,Simon P F.NatureMaterials[J], 2007, 6:992-996.
[12] Yi Z,Zhang P,Liu C,etal.Macromolecules[J],2016, 49:3343-3351.
[13] Dhumal S S,Wagh S J,Suresh A K.JournalofMembraneScience[J],2008,325(2):758-771.
[14] Maria F,Jimenez S,Song Q,etal.NatureMaterials[J], 2016, 15:760-767.
[15] Jiang Ming(江 明),Eisenberg A,Liu Guojun(刘国军),etal. Supramolecular Self Assembly(超分子自组装)[M]. Beijing: Science Press,2006.
[16] Decher G, Hong J D,Schmitt J.ThinSolidFilms[J], 1992, 210:831-835.
[17] Saqib J,Aljundi I H.JournalofWaterProcessEngineering[J],2016, 11:68-87.
[18] Joseph N P,Ahmadiannamini R,Hoogenboom I F,etal.PolymerChemistry[J],2014, 5:1817-1831.
[19] Cho K L,Hill A J,Caruso F,etal.AdvancedMaterials[J],2015,27(17):2791-2796.
[20] Joshi R K,Carbone P,Wang F C,etal.Science[J],2014,343(6172):752-754.
[21] Nair R R,Wu H A,Jayaram P N,etal.Science[J],2012, 335:442-444.
[22] Hung W S,Tsou C H,Guzman M,etal.ChemistryofMaterials[J],2014,26(9):2983-2990.
[23] Hu M,Mi B.EnvironmentalScience&Technology[J],2013,47(8):3715-3723.
[24] Sharath K,Bishnu P B,Harshal D,etal.AdvancedMaterials[J],2017,29:1603945.
[25] Kaushik D,Manas P,Kanhu C R,etal.JournaloftheAmericanChemicalSociety[J],2017, 139:13083-13091.
[26] Lv Yan(吕 嫣),Du Yong(杜 勇),Yang Shangjin(杨尚锦),etal.ActaPolymericaSinica(高分子学报)[J],2017, 12:1-10.
[27] Wang J,Yang H,Wu M,etal.JournalofMaterialsChemistryA[J],2017, 5:16289-16295.
[28] Femmer T,Kuehne A J,Wessling M.LabonaChip[J],2014, 14:2610-2613.
[29] Femmer T,Flack I,Wessling M.ChemieIngenieurTechnik[J],2016.
[30] Low Z X,Chua Y T,Ray B M,etal.JournalofMembraneScience[J],2016, 523:596-613.
[31] Cheng L,Zhang P B,Zhao Y F,etal.JournalofMembraneScience[J],2015, 492:380-391
[32] Cheng L,Zhu L P,Zhang P B,etal.JournalofMembraneScience[J],2016, 497:472-484.
[33] Ng L Y,Mohammad A W,Ng C Y.AdvancedinColloidandInterfaceScience[J],2013, 197/198:85-107.
[34] Lee H,Dellatore S M, Miller W M,etal.Science[J],2007,318(5849):426-430.
[35] Jiang J,Zhu L,Zhu L,etal.Langmuir[J],2011, 27:14180-14187.
[36] Fang L,Shi J,Zhu B,etal.JournalofMembraneScience[J],2013, 448:143-150.
[37] Shi J,Fang L,Li H,etal.JournalofMembraneScience[J],2013, 437:160-168.
[38] Zhang Peibin(张培斌),Tang Anqi(唐安琪),Lu Jingyu(路景驭),etal.JournalofFunctionalPolymers(功能高分子学报)[J],2017,30(1):1-14.
[39] Li B,Liu W,Jiang Z,etal.Langmuir[J],2009, 25:7368-7374.
[40] Zhang R,Su Y,Zhao X,etal.JournalofMembraneScience[J],2014, 470:9-17.
[41] Zhao J,Su Y,He X,etal.JournalofMembraneScience[J],2014, 465:41-48.
[42] Wang Y Q,Wang T,Su Y L,etal.Langmuir[J],2005, 21:11856-11862.
[43] Hester J F,Banerjee P,Mayes A M.Macromolecules[J],1999, 32:1643-1650.
[44] Hester J F,Banerjee P,Won Y Y,etal.Macromolecules[J],2002, 35:7652-7661.
[45] Zhu L P,Zhu B K,Xu L,etal.ColloidsandSurfacesB:Biointerfaces[J],2007, 57:189-197.
[46] Yi Z,Zhu L P,Xu Y Y,etal.JournalofMembraneScience[J],2011,385:57-66.
[47] Zhao X T, Chen W,Su Y L,etal.JournalofMembraneScience[J],2013, 441:93-101.
[48] Ying Y,Ying W,Li Q,etal.AppliedMaterialsToday[J],2017, 7:144-158.
