Original Research Paper Pulmonary delivery of liposomal dry powder inhaler formulation for effective treatment of idiopathic pulmonary fibrosis
2018-03-12ChennkesvuluMishrASudheerSowmySuryprkshReddyBhrgv
S.Chennkesvulu*,A.MishrA.Sudheer,C.Sowmy,C.Suryprksh Reddy,E.Bhrgv
harmacy Department,NDDS Laboratory,Donors Plaza,Faculty of Technology and Engineering,The Maharaja Sayajirao University of Baroda,Gujarat,India
harmacology Department,Faculty of Technology and Engineering,The Maharaja Sayajirao University of Baroda,Gujarat,India
enter for Pharmaceutical Research,Pharmaceutics Department,Raghavendra Institute of Pharmaceutical Education and Research,Anantapuramu 515721,Andhra Pradesh,India
1. Introduction
Idiopathic pulmonary fibrosis(IPF)is a chronic,progressive fi brosing interstitial lung disorder associated with histopathologic and/or radiologic pattern of unknown etiology[1].Conventional management of patients with IPF has been primarily based on the concept that suppressing in fl ammation would prevent progression to fibrosis.Although the pathogenesis is incompletely understood,IPF is a disease of abnormal wound repair and remodeling in the lung rather than an infl ammatory disease.Therefore,treatment strategies are no longer intended at reducing in fl ammation,but rather at preventing or inhibiting the fi broproliferative responses and enhancing ef fi cient alveolar epithelial repair.Budesonide,an Inhaled corticosteroid(ICS)is most commonly used in the treatment of idiopathic pulmonary fibrosis.Second most commonly used drug to prevent fibrosis is colchicine[2].Rapid clearance from the site of action and systemic side effects are some of the problems associated with single drug treatment.These problems can be avoided by combined drug treatment and selection of delivery system which delivers the drug into the lungs locally for a longer period of time and avoids systemic exposure of the drug.This is a new treatment that combines ICS(budesonide)and anti- fi brotic agent(colchicine)in a single inhaler.The ef fi cacy and safety of the individual components are well established.By combining two medications we can treat in fl ammation to prevent attacks and at the same time open up constricted airways to ease breathing and also to reduce the risk of potentials it affects that could arise from taking a single drug at a higher dosage.
Liposomes can prolong the duration of drug exposure,acting as a slow-release reservoir.Liposomes are arti fi cial lipid vesicles composed of concentric lipid bilayers that alternate with aqueous compartments[3].The liposomal encapsulation has been shown to reduce the entry of the agent into the systemic circulation,compared with free drug and provide distribution throughout the airspace of the lung.The resulting decrease in the frequency of drug dosing will signi fi cantly improve the quality of life and at the same time reduce health care cost.Well-designed dry powder inhalers are highly ef ficient systems for pulmonary drug delivery.DPI system that will allow inhalation administration of all drugs is presently delivered with MDIs.With constrain of the propellant phase out and short-term stability of liposomal aqueous dispersion the most viable alternative would be to deliver the liposomal encapsulated drug in dry form.It was hypothesized that liposomal BUD and COL will control their release rate of the drug for a longer duration at the localized site and is expected to reduce systemic side effects and frequency of dosing.
In the current study,liposomal dry powder inhaler formulation comprising of Budesonide and Colchicine was formulated to achieve site speci fi c targeted delivery with prolonged residence time in the lungs and to achieve maximum respirable fraction with improved dispersibility which may play a promising role in effective management of idiopathic pulmonary fibrosis.
2. Materials and methods
2.1. Materials
Budesonide(BUD)and Colchicine(COL)samples were given by Sun Pharma Ltd(Vadodara)and Zydus Cadila(Ahmedabad).Cholesterol(CHOL)was purchased from S.D.Fine Chemicals,India.1,2-Dipalmitoyl-snglycero-3-phosphoglycerolsodium(DPPG),Hydrogenated Soyaphosphotidylcholine(HSPC)and Soyaphosphatidylcholine(SPC)were gift samples from Lipoid,GmbH,UK and G enzyme,Switzerland.
2.2. Methods
2.2.1. Preparation of liposomes
2.2.1.1.Preparation of BUD liposomes byTFH method.Liposomes of BUD consisting of DPPG,DPPC,HSPC,EPC,SPC and CHOL were prepared by TFH technique[4].The lipids and BUD were dissolved in a mixture of chloroform and methanol(ratio 2:1 v/v)in a 250 ml round bottom f l ask in different molar ratios.The solvent was evaporated by rotary f l ash evaporator.The thin dry,lipid film thus formed was hydrated using aqueous hydrating medium(distilled water)at 60°C.The formed liposomal dispersion was sonicated(3 cycles,60%amp,0.6 cycle)using probe sonicator.Resultant Liposomes were subjected to centrifugation at 6000 rpm,4°C for 10 min using an ultra-cooling centrifuge.The liposomal suspension was decanted and drug pellet was separated.BUD:lipid ratio(1:17.5)with DPPG:HSPC:CHOL(4:5:1)formed uniform dispersion without f l aking,selected for further optimization to achieve maximum drug entrapment which was stored in amber colored vials,purged with nitrogen,sealed and stored in the refrigerator until further use.
2.2.1.2.Preparation of COL liposomes byTFH method.Liposomes of COL consisting of SPC,DPPG and CHOL were prepared by TFH technique[4].The liposomes were prepared by the procedure described above and the resultant liposomal suspension was passed through the Sephadex-G50 column and analyzed for drug content.COL:lipid ratio(1:17.5)with DPPG:SPC:CHOL(3:6:1)formed uniform dispersion without f l aking,selected for further optimization to achieve maximum drug entrapment which was stored in amber colored vials,purged with nitrogen,sealed and stored in the refrigerator until further use.
In this investigation,we have used sonication technique to achieve liposomes of nano size range.The unentrapped drug from liposomes was separated by controlled centrifugation at low-speed technique and passing the liposomal suspension through Sephadex column.Brief l y,the size of liposomal dispersion was reduced by sonicating liposomal dispersion(preheated to 60°C using thermostat)in probe sonicator.The unentrapped drug was removed from the liposomal suspension by centrifugation at 6000 rpm for 10 min at a 4°C temperature for BUD and the liposomal suspension was passed through the Sephadex-G50 column for COL.The liposomal dispersion was decanted and analyzed for entrapped drug content.Drug pellet was dissolved and analyzed for unentrapped drug content.The liposomal dispersion of BUD and COL thus obtained was filled in an amber colored vial under a nitrogen atmosphere,sealed and stored in the refrigerator until required for further experiments.
2.2.2. Analytical methods for determination of drug entrapment efficiency and in vitro drug diffusion studies
The analytical methods for chemical characterization of liposomal formulations for the determination of the drug entrapment efficiency and in vitro drug diffusion studies have been developed considering the reference methods reported by the Prasad AVSS et al.[5].The analytical method for drug entrapment efficiency have been developed by preparing aliquots of standard stock solution of Budesonide(1 mg/ml)and Colchicine(1 mg/ml)were transferred separately to 10 ml volumetric f l asks and were diluted up to the mark with methanol.The absorption maxima(λmax)were determined by scanning12 µg/ml solution(Budesonide)and 6µg/ml(Colchicine)against methanol as reagent blank on UV-Visible Spectrophotometer(UV-1700,Shimadzu).The absorption of all the prepared Budesonide and Colchicine solutions was then measured at the absorbance maxima,243 nm and 243.2 nm respectively against the reagent blank.The readings were recorded in triplicate.The analytical method for in vitro drug diffusion studies have been developed by preparing aliquots of standard stock solution of Budesonide(1 mg/ml)and Colchicine(1 mg/ml)were transferred separately to 10 ml volumetric f l asks and were diluted up to the mark with PBS pH 7.4 with 1%SLS.The absorption maxima(λmax)was determined by scanning 12 µg/ml solution(Budesonide)and 6µg/ml(Colchicine)against PBS pH 7.4 with 1%SLS as reagent blank on UV-Visible Spectrophotometer(UV-1700,Shimadzu).The absorption of all the prepared Budesonide and Colchicine solutions was then measured at the absorbance maxima,245.6 nm and 245.5 nm respectively against the reagent blank.The readings were recorded in triplicate.
The interference and interaction studies between drug and excipients were also performed.The solution containing the same concentration of the drug in the presence of the different excipients individually and in combination has been taken to detect any change in the spectra of the known concentration solutions of Budesonide(12µg/ml)and Colchicine(6µg/ml).
2.2.3. Liposome characterization
2.2.3.1.Liposome size.The size of liposomes was measured by dynamic light scattering with a Malvern Zeta Sizer 3000 HS(Malvern Instruments,Malvern,UK).Diluted liposomal suspension was added to the sample cuvette and then cuvette is placed in zeta sizer.The sample was stabilized for 2 min and reading was noted.The average particle size was determined by performing the experiments in triplicate[6].
2.2.3.2.Zeta potential.The zeta potential of developed liposomes was determined using Malvern Zeta sizer 3000 HS(Malvern Instruments,Malvern,UK)[7].
2.2.3.3.Drug entrapment(DE).To determine DE,free and entrapped drug was measured.But in the case of COL,there is no possibility to determine the free drug content.The free BUD(un-entrapped)in the liposomal dispersion was separated by controlled centrifugation at low-speed method.Brief l y,the liposomal dispersion was centrifuged at 6000 rpm,4°C for 10 min using sigma centrifuge and the liposomal dispersion was removed without disturbing the drug pellet.The drug pellet was dissolved in methanol and estimated for unentrapped drug content.A fixed volume of liposomal suspension was withdrawn and dissolved in methanol and estimated for entrapped drug content[8].
2.2.3.4.Content uniformity test.Accurately weighed 25 mg,40 mg and 65 mg liposomal DPIs samples were withdrawn 10 times from the bulk were tested for content uniformity test as per USP.
2.2.4. Freeze drying of mixture of BUD and COL loaded liposomes
Freeze drying technique was used for stabilization and to achieve desired properties for pulmonary administration.BUD and COL liposomes prepared by the optimized process and formulation components.Mixture of BUD:lipid ratio(1:17.5)(equivalent to 10 mg of BUD)composed of DPPG:HSPC:CHOL(4:5:1)and COL:lipid ratio(1:17.5)(equivalent to 12 mg of COL)composed of DPPG:SPC:CHOL(3:6:1)was freeze dried with different cryoprotectants and 10%glycine as anti-adherent to preserve the vesicular size and shape.The resultant dispersion was frozen overnight at−40 °C,dried for 48h at 50 m bar using a HetoFreeze-dry system(Hetodry,Denmark).The effect of the use of different cryoprotectants and anti-adherent on particle size,PDR,and in vitro performance among developed Liposomal DPI was studied.The porous cake thus formed was sized successively through#120 and#240 sieves.PDR(Percent drug retained)of freeze dried liposomes was determined following dehydration-rehydration cycle.
2.2.5. Sem photomicrographs
Samples were attached to sample stubs using double-sided tape and then viewed using an accelerating voltage of 15 kilovolts at the magnification of 2500x to 5000x by Scanning electron microscopy(JSM-5610LV,JEOL,Japan).
2.2.6. Liposomal DPI characterization
The Liposomal DPIs were characterized for the following physicochemical properties.
2.2.6.1.Angle of repose.The pile of powder was carefully built up by dropping the powder material through a glass funnel tip from height of 2 cm.The angle of repose was calculated by inverting tangentially the ratio of height and radius of the formed pile.Angle of repose was performed in sixtuplicate[9].
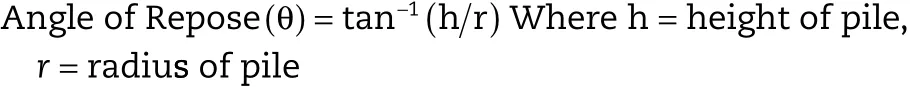
2.2.6.2.Residual water content.The residual moisture content of the DPI formulations was determined by using Karl-Fischer titrator(Aqua-Cal,Analab Instruments,Vadodara,India).The commercially available pyridine-free reagent was standardized with a known quantity of water(5µl)and the volume of Karl-Fischer reagent required for neutralizing the water was used for calculating “Water Equivalent Factor”(WEF).The moisture content present in the formulation was determined by adding 50 mg of DPI formulation in the titrator and of KF reagent required for neutralizing the water present in the formulation.
2.2.6.3.Rehydrated liposomal particle size.The Liposomal DPI formulations were rehydrated and diluted with distilled water.The dispersion was filled in sample cuvette and analyzed for particle size by laser diffraction using the Malvern particle size analyzer(Malvern Zeta sizer 2000 SM,U.K.).
2.2.7. Characterization of aerosol performance
The in vitro pulmonary deposition was evaluated using an eight stage,nonviable Andersen cascade impactor with a preseparator(Graseby-Andersen,Atlanta,GA,USA).A cascade impactor was used to collect data on powder performance.Brief l y,eight filters were placed on collection plates which were housed within eight air tight stages arranged serially and stacked on a level setting.The air was then pumped through the stages at a rate of 28.3 lit/min via a vacuum pump and samples were introduced at the top of the impactor device.The powder was allowed to deposit among the stages for 10 seconds,after which air f l ow was stopped.Cut-off particle aerodynamic diameters for each stage were provided by the manufacturer as follows:Pre-separator–10.00µm,stage 0–9.00µm,stage 1–5.8µm,stage 2–4.7µm,stage 3–3.3µm,stage 4–2.1µm,stage 5–1.1µm,stage 6–0.7µm,stage 7–0.4µm and the final stage(stage 8)is intended to collect any remaining particulates,though complete entrainment is nearly impossible.A size “2”hard gelatin capsule(Universal capsules,Mumbai,India)was filled with 25 mg,40 mg and 65 mg of powder equivalent to 300µg of BUD,600µg of COL and M2(300µg of BUD and 600µg of COL)aerosolized using Rotahaler(Cipla Ltd.,Mumbai,India).Ten capsules were actuated for each impaction with each capsule for 10 secs.The drug deposited in different parts such as induction port,preseparator,individual impaction plates,powder remaining in the capsule and inhaler device was rinsed with methanol.From drug deposition data the emitted dose,fine particle dose,fine particle fraction,mass median aerodynamic diameter and geometric standard deviation were calculated[10].
2.2.8. In vitro diffusion studies
Dialysis membrane(250-9U,molecular weight cut off:12000 Dalton;Sigma,Hyderabad,India),200µm in thickness,pH 5.8 to 8,breaking strength 2.75 kg f/cm and porosity 0.45µm was used as an artificial membrane for preliminary in vitro studies because of simplicity,homogeneity and uniformity.This membrane was pre-treated with ethanol(95%v/v)followed by hydration in pH 7.4,phosphate buffer saline(PBS)with 1%w/v SLS and 1 mM EDTA for 24 h prior to permeation studies.Receptor compartment containing 13 ml of PBS,pH 7.4 having 1%w/v SLS,with constant stirring simulated highly perfused pulmonary condition.Diffusion studies were carried out for plain drug DPIs(BUD,COL and combination of both)and developed liposomal DPIs.The study was performed by dispersing plain drug Suspensions and liposomal DPIs in 2 ml of distilled water.Formulations to be compared were separately transferred to the donor compartment and receptor compartment,stirred at 50 rpm.1 ml of the sample was withdrawn from the receptor compartment at definite time intervals and an equivalent amount of fresh medium was replaced to the receptor compartment.All diffusion studies and sample analysis were carried out three times and mean values along with standard error of the mean were calculated.
2.2.9. In vivo studies
All experiments and protocols described in the present study were approved by the Institutional Animal Ethics Committee(IAEC)of M.S.University,Baroda and with permission from Committee for the Purpose of Control and Supervision of Experiments on Animals(CPCSEA),Ministry of Social Justice and Empowerment,Government of India.Healthy adult male rats weighing 200–250 g(generously provided by Sun Pharmaceuticals,Vadodara,India)were used for the study.The animals were housed in according to CPCSEA guidelines.
2.2.9.1.Experimental design.48 Healthy adult maleWistar rats were randomly divided into four groups(12 each)as follows:
Group I:Control rats treated intratracheally at a dosage with 0.25 ml of 0.9%NaCl.
Group II:Received with Bleomycin(2.5 U/kg dissolved in 0.25 ml of 0.9%NaCl)a single sublethal dose by intratracheally[11].
Group III:Bleomycin single dose+Plain drug(BUD 0.5 mg/kg body weight(bw)and COL 1.5 mg/kg bw,intratracheally)was administered on Day 1 and Day 8 of 14 d protocol[12].
Group IV:Bleomycin single dose+LDPI formulation(BUD 0.5 mg/kg bw and COL 1.5 mg/kg bw,intratracheally)was administered on Day 1 and Day 8 of 14 d protocol.
2.2.9.2.Induction of idiopathic pulmonary fibrosis.To produce pulmonary fibrosis,animals received endotracheally,by the transoral route,a single sublethal dose of bleomycin(2.5 U/kg-1 dissolved in 0.25 ml of 0.9%NaCl).Control animals were subjected to the same protocol but received the same volume of intratracheal saline instead of bleomycin.Tracheal instillation was carried out under Ketamine hydrochloride and Diazepam(60 mg/kg and 5 mg/kg)anaesthesia.
2.2.9.3.Intratracheal administration of tested drugs.Before intratracheal administration of test drugs,the rats were anaesthetized with Ketamine hydrochloride(60 mg/kg,i.p)and Diazepam(5 mg/kg,i.m).Hair was shaved at neck region and the area was disinfected with spirit.The trachea was exposed by blunt dissection of the sternohyoideus muscle and a small midline incision was made over the trachea.The specified dose of Plain drug(BUD and COL)and DPIs(Mixture of BUD and COL)were suspended separately in prewarmed saline and was slowly instilled into trachea over a 1 min period with syringe using as a 20-guage needle.Following instillation,the wound was closed with 3-0 Dexon sutures.Intraperitoneal Ampicillin(10 mg/kg)and Diclofenac Sodium(1 mg/kg)were administered to the rats for every 3 d to combat infection and pain of the animals.Following anesthesia,the animals were allowed to recreate under the heating lamp.After recovery,animals were housed in individual plastic cages with free access to food and water for the remainder of the study.The above protocol was followed on Day 1 and Day 8 of 14 d study[13].
2.2.9.4.Bronchoalveolar lavage(BAL)for analysis of inf l ammatory cell profile.BAL was performed at day 7 and 14 after intratracheal instillation of bleomycin.The differential count was determined from a minimum of 300 cells.Total cell counts were made in a haemocytometer[14].
2.2.9.5.Biochemical measurements.
2.2.9.5.1.Hydroxyproline(HPO)determination in lung tissue.The HPO activity was measured in the lung tissue.The removed right lung of the animal was homogenized in 2 ml of phosphate-buffered saline(PBS,pH 7.4)and then digested in 6N HCL for 8 h at 120°C.Five microliters of sample were mixed with 5 µl citrate/acetate buffer and then 100 µl of Ehrlich’s solution before incubated for 15 min at 65°C.The hydroxyl proline concentration was analyzed by spectrophotometer at 550 nm[13].
2.2.9.5.2.Myeloperoxidase activity(MPO)determination in lung tissue.The MPO activity was measured in the lung tissue.Tissue samples were homogenized in 50 mM potassium phosphate buffer(PB,pH6.0),and centrifuged at 41,400×g(10 min);the pellets were suspended in 50 mM PB containing 0.5%hexadecyl trimethyl ammonium bromide.After three freeze and thaw cycles with sonication between cycles,the samples were then centrifuged at 41,400×g for 10 min.Aliquots(0.3 ml)were added to 2.3 ml of a reaction mixture containing 50 mM PB,o-dianisidine,and 20 mM H2O2solution.One unit of enzyme activity was defined as the amount of the MPO present which caused a change in the absorbance measured at 460 nm for 3 min.The results are expressed as U/g tissue[15].
2.2.9.6.Histological assessment.Histological grading of lesions was performed for extent and severity of in fl ammation and fibrosis in lung parenchyma.Brie fl y,three lung sections from each animal were systematically scanned and each successive field was scored[11].
2.2.10.Stability studies
Comparative stability studies were carried out for the potential liposomal DPI formulations at room conditions(25°C±2°C,60%±5 RH)and at refrigerated conditions(2-8°C)up to six months.Liposomal DPI formulations containing 300µg BUD,600µg COL and a mixture of 300µg BUD and 600µg COL(M2)were filled into gelatin capsule shells(Size 2)and were packed in HDPE bottles(silica bags as dehumectant)under nitrogen cover and the bottle was sealed with PVC coated aluminum foil.The liposomal DPIs formulations were also examined visually for the evidence of caking and discoloration.The content of the capsules is tested for assay,degradation,water content,PDR,emission and FPF.
3. Results and discussions
Liposomes of BUD and COL were prepared by the selectedTFH method using DPPG,HSPC,SPC and CHOL that are optimized to achieve maximum PDE and desired size range.
3.1. Optimization of TFH method process variables
The organic solvent system,chloroform:methanol(2:1)was used for dissolving the formulation components like HSPC,DPPG,SPC,CHOL and drug.Process variables,such as vacuum conditions for dry film formation,hydration time and speed of rotation of f l ask were optimized for desired results.The effect of one variable was studied at a time keeping other variables constant.Variables like hydration time and speed of rotation of f l ask were kept constant to optimize vacuum conditions for dry film formation.The vacuum of 600 mm of Hg for 50 min for BUD and 60 min for COL was found to be optimum for complete evaporation of solvent mixture and producing more translucent and thin lipid film.Variables,vacuum conditions for dry film formation and hydration time were kept constant to optimize speed.A speed of 100 rpm was found to be adequate to give the thin,uniform and completely dry film.Hence,100 rpm speed,rotation of f l ask was selected to be optimum for liposomal preparation.Variables,vacuum conditions for dry film formation and speed were kept constant to optimize hydration time.Hydration time(1h)was found to be optimum for both BUD and COL liposome preparation.
BUD:lipid ratio–Increase in the lipid proportion relative to drug led to the increase in the drug entrapment from 23.07±0.65%(BB 1:7.5)to 74.22±0.97%(BH 1:17.5),increase in drug entrapment efficiency is compensated with proportionate increase in lipids i.e.to use more lipids to entrap constant amount of drug.Hence,drug:lipid ratio of BH 1:17.5 with DPPG:HSPC:CHOL(4:5:1)was selected due to acceptable drug entrapment(74.22±0.97%)of BUD.(Table 1)
COL:lipid ratio–Increase in the lipid proportion relative to drug led to the increase in the drug entrapment from 19.16±1.34%(CB 1:5)to 50.94±2.04%(CG 1:17.5),increase in drug entrapment efficiency was compensated with proportionate increase in lipids i.e.to use more lipids to entrapconstant amount of drug.Hence,drug:lipid ratio of 1:17.5 with DPPG:SPC:CHOL(3:6:1)was selected due to acceptable drug entrapment efficiency(50.94±2.04%)of COL(Table 2).Increased retention of drug in liposomes over the longer period of time with an increase in cholesterol content may be due to increased bilayer stability by incorporation of cholesterol into the bilayers.Batches BH and CG retained drug for a longer period of time,hence selected for development of liposomal dry powder inhaler formulations.

Table 1–Optimization of liposomal BUD Formulation by TFH method.
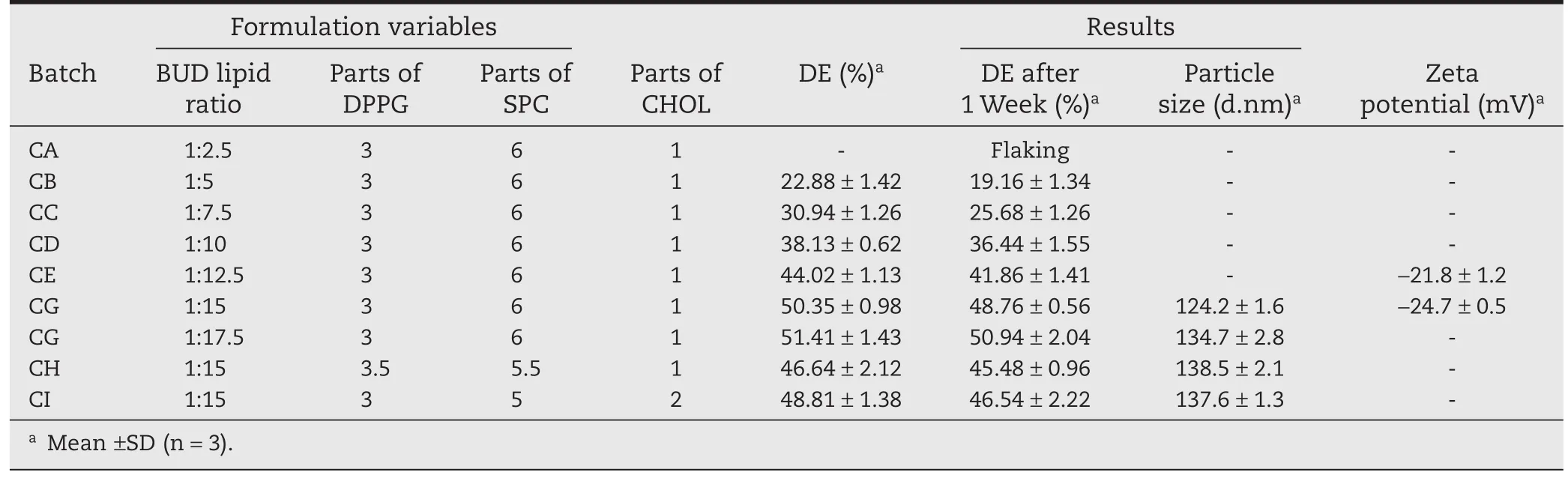
Table 2–Optimization of liposomal COL Formulation by TFH method.
3.2. Analytical methods for determination of drug entrapment efficiency and in vitro drug diffusion studies
It has been observed that no interference found with excipients.UV-spectra of BUD at λ max=243.2 nm and UV-spectra of COL at λ max=243.0 nm in methanol are suitable to calculate the amount of drug entrapment in formulation development and at the same time UV-spectra of BUD at λ max=245.6 nm and UV-spectra of COL at λ max=245.5 nm in PBS pH 7.4 with 1%SLS are suitable to calculate the amount of drug release in diffusion studies.Hence these methods can be used for individual estimation of BUD and COL to calculate drug entrapment during formulation development and drug release studies during diffusion studies respectively.
3.3. Characterization of liposomes
The formulated Liposomes were characterized for physicochemical properties and the results were tabulated in(Table 3),(Figs.1&2).
3.4. Liposomal DPI characterization

Fig.1–Unsonicated Liposome Photograph in Olympus Microscope(40X).

Table 3–Physicochemical characterization of Liposomes.

Fig.2–SEM image of batch M2 liposomal DPI.
The particle size of Liposomal DPI formulation(M2)found to be 0.755±1.2µm,which would be beneficial for deep lung deposition by the process of diffusion(Brownian motion).Batch B2(97.8±0.5%),C2(97.9±0.8%)and M2(98.6±1.0%)showed highest PDR compared to all other prepared formulations(Table 4).Batch B2,C2 and M2 showed the angle of repose in the range 30-350.Batches prepared with Mannitol as cryoprotectant showed the least moisture(2.8±0.4%)in comparison with batches prepared with Lactose and Sucrose.Mannitol was found to be effective in maintaining the size of liposomal vesicles.All the samples were within the limits as per USPXXVII and passes the content uniformity test.
3.5. Characterization of aerosol performance
Percentage FPF is the major criterion,which defines the eff icacy of the DPI in lungs.Higher the%FPF,higher will be the deposition of DPI in lungs.The%FPF values of the liposomal DPI formulations prepared using mannitol as cryoprotectant were found to be highest than prepared using lactose and sucrose.%Emission is the criterion,which shows the amount of DPI emitted from the inhalation devices.More will be the%Emission,better will be the DPI deposition and less will be the loss in the device.%Dispersibility is a criterion for determination of f l ow properties of the powder from the inhalation device.Higher%dispersibility was recommended for good f l ow of powder from the inhaler.The MMAD value of Batch M2 found suitable for efficiently targeting the airways.Batch M2 due to low residual water content which improved aerosol dispersion performance showed higher%FPF,more%Emission,higher%dispersibility(good f l ow),good MMAD(Table 5).
3.6. In vitro diffusion studies
Regression analysis[BUD(R2=0.990),COL(R2=0.999)]of the data proved the linearity of plots in the concentration range used.The interference of formulation components was checked and found non-interfering at absorbance maxima of BUD and COL.It was found that prepared liposomal DPIs sustained the drug release up to 24 h or more.There are two rate-controlling barriers inf l uencing the drug diffusion to the receptor compartment,one is the liposomal membrane and the other is the artificial membrane.The percentage drug diffusion of liposomal drugs is found to be dependent upon the composition DPI formulations.Hence,we can conclude that the liposomal membrane controls the drug diffusion and not the artificial membrane.The artificial membrane acts only as a physical barrier preventing the liposomes to diffuse into the donor compartment and notregulating the drug diffusion to the receptor compartment.Liposomal encapsulation,the composition of liposomal membrane and charge are expected to help in retaining the drug within the lung.Hence,we can conclude that the liposomal membrane controls the drug diffusion and not the artificial membrane.All these observations lead us to the conclusion that liposomal DPI formulations delivery has a greater potential for sustained diffusion of the drug.Drug diffusion from liposomal DPI formulations obeys Higuchi’s diffusion controlled model and the diffusion rate is close to first order kinetics.The diffusion rate depends upon the physicochemical property,concentration of drug within the liposomes and the composition of the liposomal membrane.Hence by altering the composition of the liposomal membrane,different loading dose followed by maintenance dose can be achieved.This model of diffusion study may be used to assess the desired diffusion pattern by modulating the composition of the bilayer membrane and evaluating the formulation’s in vitro before going for in vivo.For optimized formulation(M2)cumulative percent,drug diffusion was plotted against time(t),illustrated in Fig.3 for both the drugs.
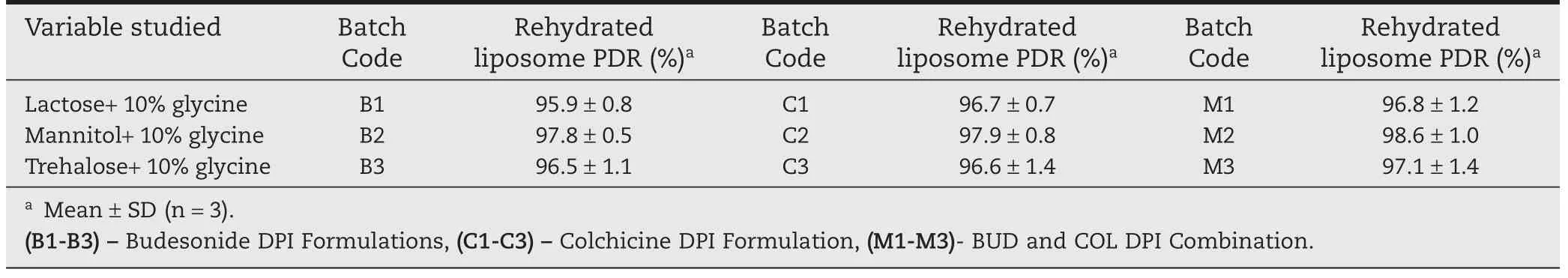
Table 4–Inf l uence of different cryoprotectants on freeze dried Liposomal DPI.
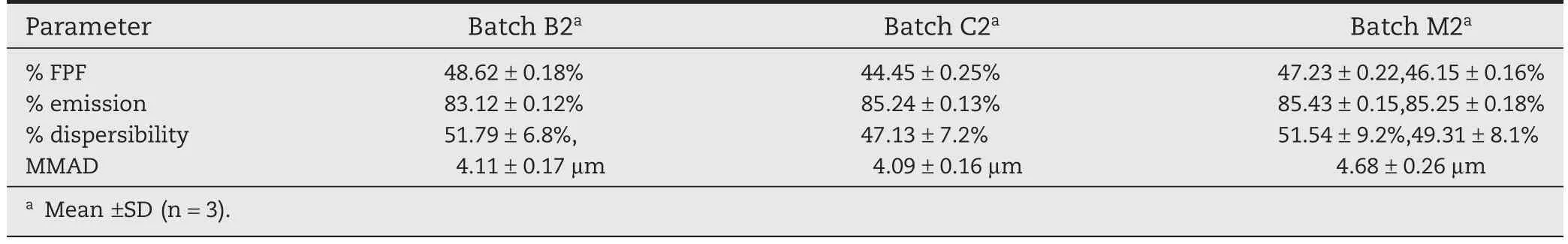
Table 5–Characterization of Aerosol performance.
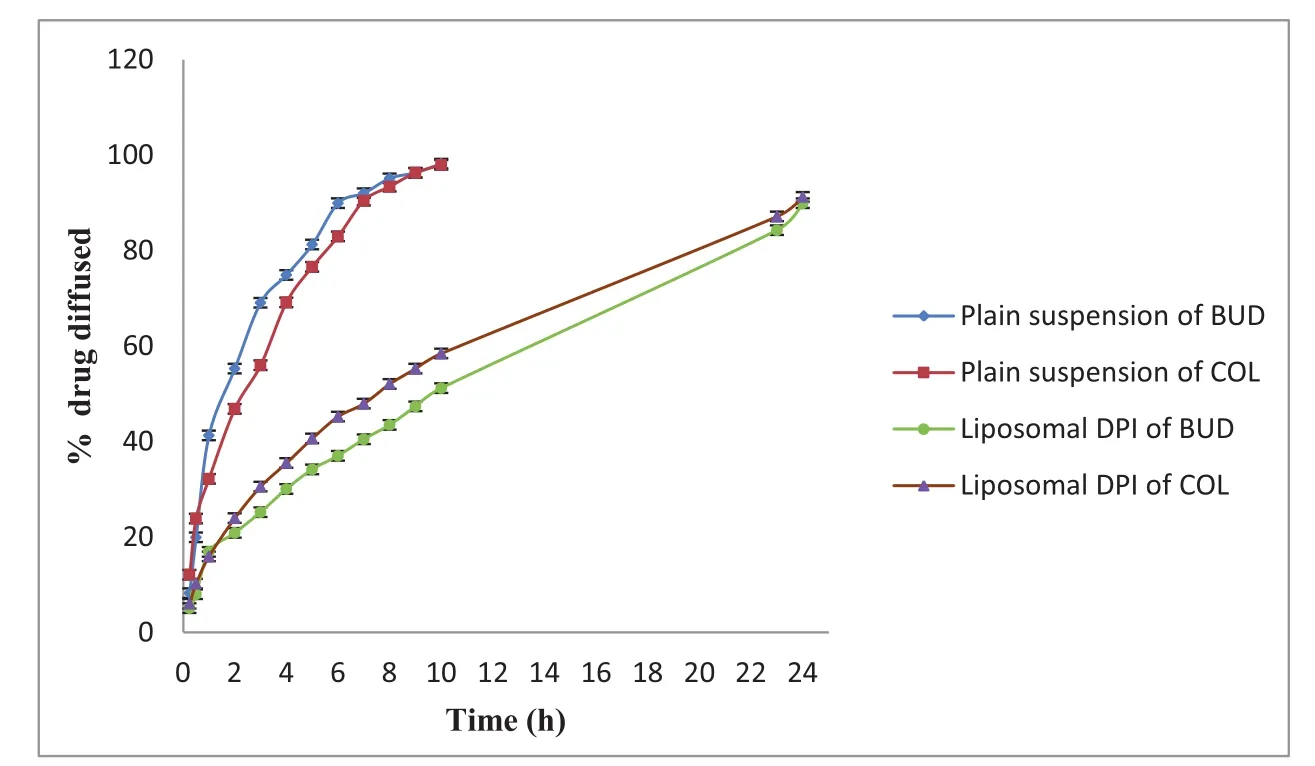
Fig.3–Comparative in vitro drug diffusion studies of optimized(M2)drug liposomal DPI formulation(mean±SD,n=3).
3.7. In vivo results
Bleomycin treatment produced a significant increase in the total cell count in the BAL f l uid as compared to the saline-treated rats and to rats that were not exposed to bleomycin(control rats).The differential cell count showed that neutrophils were markedly augmented in lungs of rat exposed to bleomycin and this augmentation was significantly prevented by the treatment of DPI formulation,and treatment with DPI formulation caused a significant reduction in the number of neutrophils from 58.56 ± 4.5(×103/ml)to 2.4 ± 0.6(×103/ml)after 14 d treatment shown in Table 6.After 14 d,the hydroxyproline content was significantly decreased from 15.84±0.65 mg/gm to 9.21±0.92 mg/gm,indicating a beneficial effect of prepared formulation against bleomycin-induced pulmonary fibrosis.After 14 d,the Myeloperoxidase activity was significantly decreased from 4.6±0.24 mg/gm to 1.3±0.12 mg/gm,indicating a beneficial effect of prepared formulation against bleomycininduced pulmonary fibrosis(Fig.4).
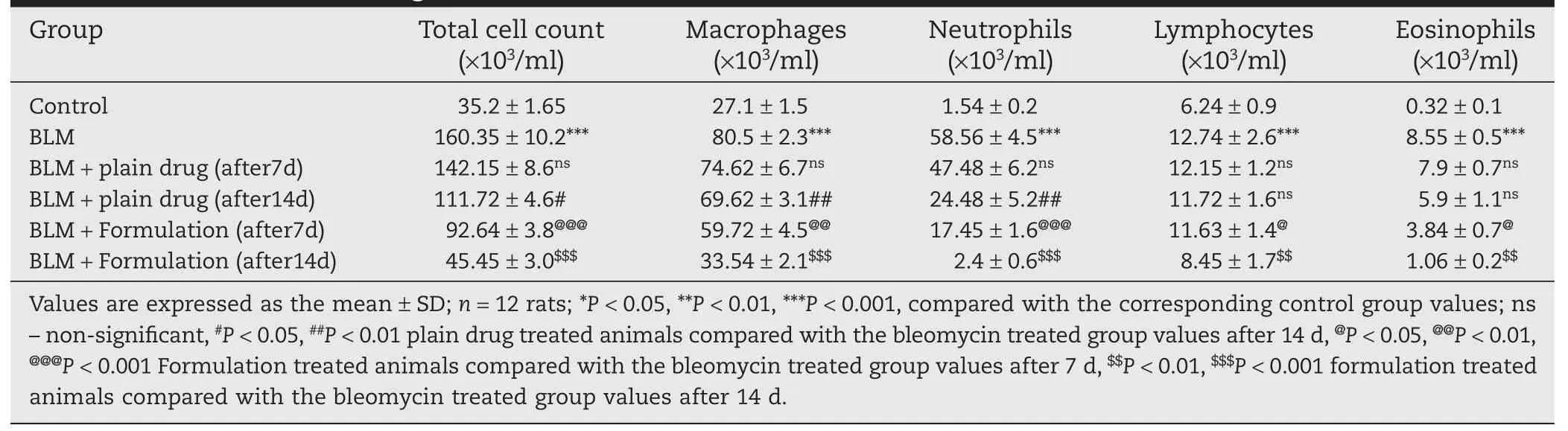
Table 6–Effects of Intra tracheal instillations of DPI formulation on BLM induced changes in total and differential cell counts of Bronchoalveolar lavage f l uid.*,**
3.8. Histopathology
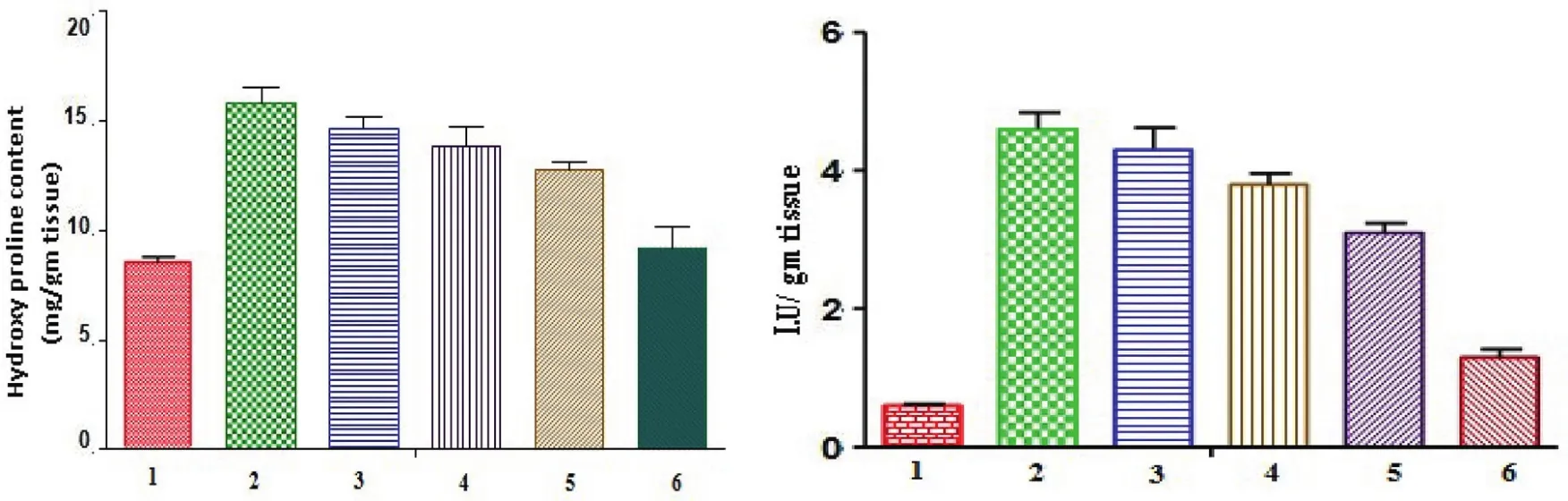
Fig.4–Hydroxyproline content and Myeloperoxidase activity of lung tissue in different treatment groups.1-Control;2–BLM***;3-BLM+Plain drug(after 7 d)ns1-Control;2–BLM***;3-BLM+Plain drug(after 7 d)*;4-BLM+Plain drug(after 14 d)*4-BLM+Plain drug(after 14 d)**5-BLM+Formulation(after 7 d)*5-BLM+Formulation(after 7 d)***6-BLM+Formulation(after 14 d)**6-BLM+Formulation(after 14 d)***Where,ns–non-significant,*P<0.05,**P<0.01,***P<0.001,compared with the corresponding control group values.
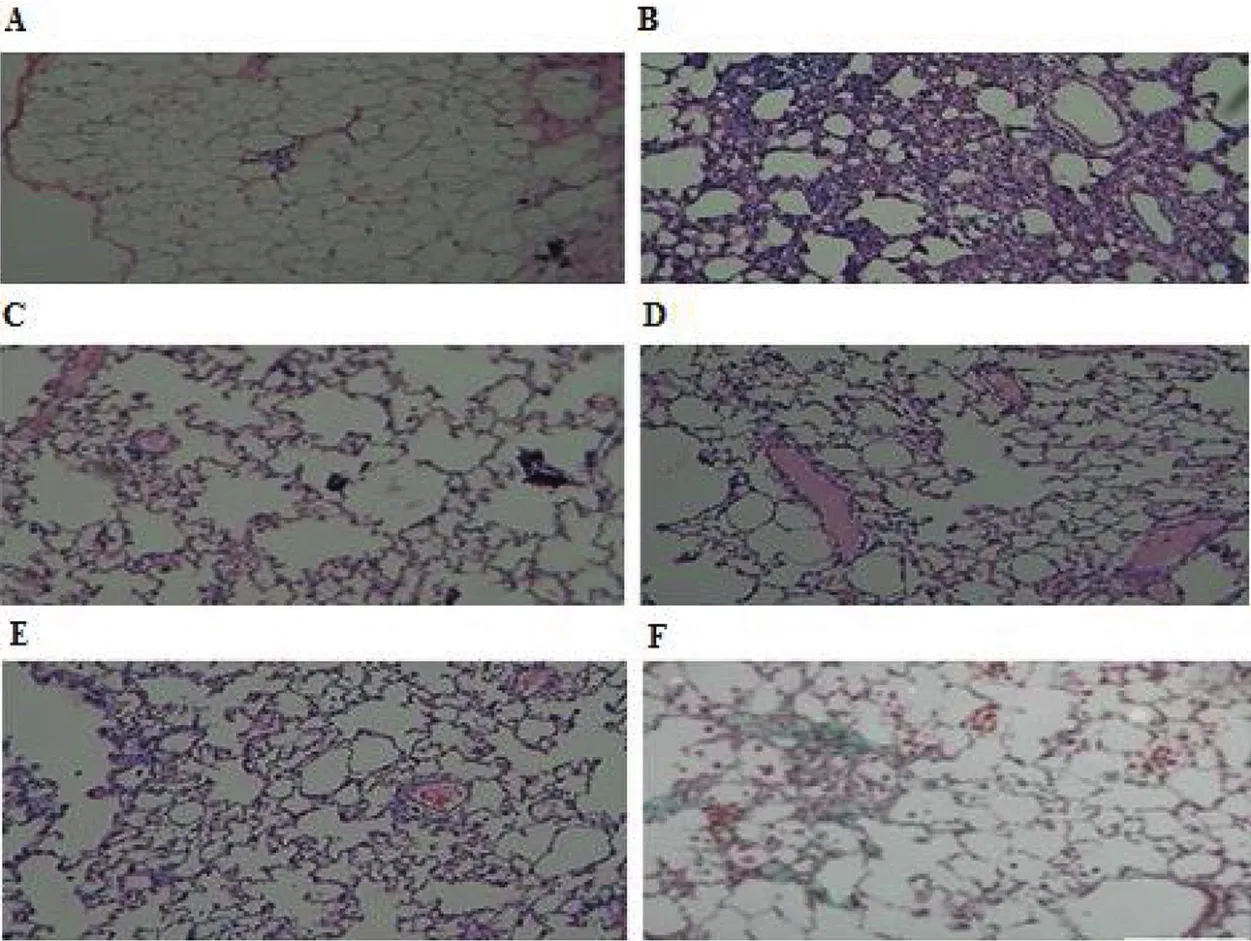
Fig.5–Histo-morphological appearances of lung samples stained with hematoxylin eosin.A–Control,B–BLM,C–BLM+Plain drug(after 7 d treatment),D–BLM+M2 DPI Formulation(after 7 d treatment),E–BLM+Plain drug(after 14 d treatment),F–BLM+M2 DPI Formulation(after 14 d treatment).
Lung tissue samples in the Control Group had a normal pulmonary architecture with thin-lined alveolar septa,normal cellularity and only a few alveolar macrophages,while the samples in Group BLM had typical fibrotic changes including infiltration with excessive inf l ammatory cells large collagen depositions,large fibrous areas and collapsed alveolar spaces were found in the samples viewed under a light microscope.(Fig.5 A,B)After treatment with prepared plain drug and DPI formulations on bleomycin-induced fibrotic rats;DPI formulation had shown the decrease in a number of inf l ammatory cells and the thickness of the alveolar septa.(Fig.5 C,D,E,F).To confirm the effects of prepared plain drug and DPI formulations on the histopathology of BLM-induced lung injury and fibrosis,the overall grades(Table 7)of inf l ammatory and fibrotic changes in the lungs were estimated by numerical scoring.The scores of alveolitis and fibrosis in the lung sections of BLM+DPI formulation group was significantly decreased,compared with those of the BLM+Plain drug formulation.DPI formulation containing BUD and COL significantly prevented the developments of lung fibrosis produced by bleomycin,confirmed by the pathologic investigation,measurements of lung hydroxyproline content,Myeloperoxidase activity and BALF analysis.No significant differences were found in all parameters for optimized formulation(M2)at both room condition(25°C ±2°C,60%±5 RH)and at refrigerated conditions(2-8°C)up to six months.

Table 7–Grading schemes of histopathology tissues of different groups.
4. Conclusion
The studies supported that carrier based liposomal dry powder inhaler pulmonary drug delivery for treatment of Idiopathic Pulmonary Fibrosis exhibited prolonged drug retention at targeted site and reduced the systemic exposure.Hence,it is expected to maximize the therapeutic index,reduce the systemic side effects,frequency of dosing and dose,probably the cost of therapy.This investigation has only provided preliminary evidence that delivery of carrier based dry powder inhaler of anti-inf l ammatory and anti-fibrotic agents can confine drug primarily to lung using a liposomal dry powder inhaler delivery system and hence expected to treat the disease better and can also avoid the development of drug resistance.However,the role of liposomal dry powder inhaler formulations in the treatment of idiopathic Pulmonary Fibrosis can only be settled after experiments on atleast two animal species followed by extensive clinical trials.
5. Declaration of interest
The authors thankThe Principal ofThe Maharaja Sayajirao University of Baroda for providing required facilities to carry out the research work.
[1]King TE Jr,Pardo A,Selman M.Idiopathic pulmonary fibrosis.Lancet 2011;378:1949–1961.
[2]Richeldi L,Davies HR,Ferrara G,et al.Corticosteroids for idiopathic pulmonary fibrosis.Cochrane Database Syst Rev 2003;(3):CD002880.
[3]Riaz M.Stability and uses of liposomes.Pak J Pharm Sci 1995;8:69–79.
[4]New R.R.C.Characterization of liposomes.In:Liposomes:a practical approach.New York:Oxford University Press;1994.p.105–160.
[5]Prasad AVSS.Simultaneous spectrophotometric determination of formoterol fumarate and budesonide in their combined dosage form.Ind J Chem Technol 2006;13:81–83.
[6]El-Nesr OH,Yahiya SA,El-Gazayerly ON.Effect of formulation design and freeze-drying on properties of lf uconazole multilamellar liposomes.Saudi Pharm J 2010;18:217–224.
[7]Feng SS,Mu L,Win KY,et al.Nanoparticles of biodegradable polymers for clinical administration of paclitaxel.Curr Med Chem 2004;11:413–424.
[8]New RRC.Liposomes:a practical approach.New York:Oxford University Press;1990.p.242–243.
[9]Misra A,Jinturkar K,Patel D,et al.Recent advances in liposomal dry powder formulations:preparation and evaluation.Expert Opin Drug Deliv 2009;6:71–89.
[10]Meenach SA,Anderson KW,Zach Hilt J,et al.Characterization and aerosol dispersion performance of advanced spray-dried chemotherapeutic PEGylated phospholipid particles for dry powder inhalation delivery in lung cancer.Eur J Pharm Sci 2013;49:699–711.
[11]Moeller A,Ask K,Warburton D,et al.The bleomycin animal model:a useful tool to investigate treatment options for idiopathic pulmonary fibrosis.Int J Biochem Cell Biol 2008;40:362–382.
[12]Barnes CD,Eltherington LG.Drug dosage in laboratory animals:a handbook.Berkeley:University of California Press;1965.p.65–66.
[13]Zhao L,Wang X,Chang Q,et al.Neferine,a bisbenzylisoquinline alkaloid attenuates bleomycin-induced pulmonary fibrosis.Eur J Pharmacol 2010;627:304–312.
[14]Qin S,Alcorn JF,Craigo JK,et al.Epigallocatechin-3-gallate reduces airway in fl ammation in mice through binding to proin fl ammatory chemokines and inhibiting in fl ammatory cell recruitment.J Immunol 2011;186:3693–3700.
[15]Kacmaz A,User EY,Sehirli AO,et al.Protective effect of melatonin against ischemia/reperfusion-induced oxidative remote organ injury in the rat.Surg Today 2005;35:744–750.
杂志排行

Asian Journal of Pharmacentical Sciences的其它文章
- GUIDE FOR AUTHORS
- Original Research Paper Preparation of poly(lactide-co-glycolide)microspheres and evaluation of pharmacokinetics and tissue distribution of BDMC-PLGA-MS in rats
- Original Research Paper Intranasal administration of carbamazepineloaded carboxymethyl chitosan nanoparticles for drug delivery to the brain
- Original Research Paper Association between the physical stability of flurbiprofen suspension and the interaction of HPMC/SDS
- Original Research Paper Validation of kinetic modeling of progesterone release from polymeric membranes
- Original Research Paper The accelerated blood clearance phenomenon of PEGylated nanoemulsion upon cross administration with nanoemulsions modified with polyglycerin