咪唑并[1,2-a]喹喔啉衍生物的合成及其体外抗乳腺癌活性研究
2018-03-06许贻文陈晓乐许建华
林 晨, 许贻文, 柯 方,2, 陈晓乐,2, 许建华,2, 林 媚
吡咯并[1,2-a]喹喔啉衍生物是一类具有极高应用前景的含氮稠杂环化合物,一些高度选择性的环核苷酸碳酸二酯酶(cyclic nucleotide phosphodiesterase,PDE)同功酶抑制剂和NF-κB中的IκB激活酶抑制剂就含有咪唑并喹喔啉结构[1]。因其具有抗肿瘤、抗病毒和抗溃疡等广泛高效的生理、药理活性,具有较好的应用前景,越来越受研究者们重视,已逐渐成为药学和医学等领域的研究热点[2-4]。咪唑并喹喔啉的传统制备方法以2-氯喹喔啉和邻氟硝基苯为原料,经过亲核取代反应、硝基还原和环合过程制得产物[5-6]。2014年,Huang等采用铜盐催化获得了咪唑并[1,2-a]喹喔啉[7]。目前合成咪唑并[1,2-a]喹喔啉的方法受到很大限制,如操作繁琐、条件苛刻和底物难以制备等缺点。因此,发展相对高效、廉价的合成咪唑并[1,2-a]喹喔啉先导化合物的方法成为热点。
乳腺癌是一种女性常见恶性肿瘤,其发病率呈逐年上升趋势[8]。在所有乳腺癌患者中,人表皮生长因子受体2(human epidermal grouth factor receptor 2,HER2)阳性乳腺癌占20%~30%[9]。HER2是临床监测乳腺癌预后的重要指标之一,也是设计肿瘤靶向治疗药物的重要靶点。人类不断研究探索更加高效低毒的新药物,期待它对HER2阳性细胞具有高效杀伤作用。最近的研究发现,喹喔啉衍生物可对乳腺癌细胞具有较好的抑制作用[10]。
本研究探讨在微波辐射下,采用NiCl2和1,10-菲啰啉(1,10-phen)作为催化剂,通过2-氯甲基苯并咪唑和N-4-甲苯磺酰基-2-氨基碘苯为底物制备具有潜在抗肿瘤活性的咪唑并[1,2-a]喹喔啉衍生物的方法(合成线路如图1所示),研究其对HER2阴性和HER2阳性乳腺癌细胞的杀伤能力,为进一步筛选具有高抗肿瘤活性的喹喔啉做前期研究。

图1 咪唑并[1,2-a]喹喔啉化合物的合成
1 材料与方法
1.1材料
1.1.1试剂 2-氯甲基苯并咪唑和N-4-甲苯磺酰基-2-氨基碘苯及其衍生物均为美国Sigma-aldrich公司分析纯。其余试剂均为市售化学纯产品,未经进一步纯化;肿瘤细胞株 MCF-7和SKBR3细胞由福建医科大学新药研究所传代培养。
1.1.2仪器 微波合成仪(Initiator+,瑞典Biotage公司);高效液相色谱-质谱仪(LCMS-2020,日本岛津公司);核磁共振仪(AM-80,德国Bruker Biospin公司);1H NMR:400MHz (溶剂: CDCl3),13CNMR: 100 MHz (溶剂:CDCl3);全自动酶标仪(680型,美国BIO-RAD公司)。
1.2方法
1.2.1咪唑并[1,2-a]喹喔啉的合成方法 将1.2 mmol 2-氯甲基苯并咪唑和1 mmolN-4-甲苯磺酰基-2-氨基碘苯加入到干燥的10 mL长颈烧瓶中,依次加入0.1 mmol NiCl2,0.1 mmol 1,10-phen,3.0 mmol NaOH,再加入N,N-二甲基甲酰胺(DMF)溶剂3 mL。微波反应器设定120 W功率并加热至110 ℃反应30 min。停止反应后冷却至室温,减压浓缩,对所得粗产品进行柱层析得目标产物,计算产率。
1.2.2细胞培养 人乳腺癌细胞MCF-7和SKBR3细胞采用含10%牛血清的DMEM培养基、青霉素(1×105IU/L)和链霉素(50 mg/L)在37 ℃、体积分数为0.05的CO2孵育箱中培养,取对数生长期细胞用于实验。
1.2.3细胞划痕实验 采用细胞划痕实验测试对应化合物a~e对人乳腺癌细胞MCF-7的生长和迁移活性,具体步骤如下:收集MCF-7对数期细胞,调整细胞浓度,接种12孔板,每孔细胞数1×105个;种板后12 h,MCF-7完全贴壁,采用移液枪枪头在细胞上划“一”字,PBS洗涤3次,去除划痕下的MCF-7细胞;加入含待测药的2%血清的培养基(2% FBS,1%青霉素和链霉素混合液的DMEM),待测药终浓度设置为20.0,40.0,60.0,80.0 μmol/L;放入37 ℃、体积分数为0.05的CO2培养箱培养24 h后取样,检测化合物对MCF-7迁移活性的抑制。
1.2.4MTT实验 为进一步探讨此衍生物的抗肿瘤活性,采用MTT法测试化合物a~e对MCF-7和SKBR3的增殖抑制活性。具体步骤如下:收集对数期生长的MCF-7及SKBR3细胞,调整细胞悬液浓度约为5×104mL-1,接种于96孔板,每孔加入500 μL;37 ℃、体积分数为0.05的CO2培养箱孵育至细胞单层铺满孔底,并设置对照组(未添加化合物)、空白组(只加培养基),每组设3个复孔,待测药终浓度分别为5.0,10.0,20.0,40.0,80.0 μmol/L;并在37 ℃、体积分数为0.05的CO2培养箱培养48 h后,每孔加入5 g/L的MTT溶液20 μL,继续培养4 h;停止培养,吸去孔内培养液,每孔加入150 μL DMSO,至摇床上低速震荡10 min,使结晶充分溶解。采用全自动酶标仪检测570 nm处的吸光度(OD)值。
1.3统计学处理 结果采用GraphPad Prism 5 软件分析,计算药物的半数抑制浓度IC50。组间比较采用One Way Anova进行检验分析。
2 结 果
2.1产物与产率 在微波辅助条件下,通过高效且便宜的镍催化体系快速制备了咪唑并[1,2-a]喹喔啉衍生物,反应速度较常规加热条件下明显加快,提高了合成反应的产率和效率,具有操作简单、条件温和的优点。在最优反应条件下,选取一系列2-氯甲基苯并咪唑和N-4-甲苯磺酰基-2-氨基碘苯衍生物进行反应,上述催化体系对各种取代的N-对甲苯磺酰基-2-碘苯胺和2-氯甲基苯并咪唑均具有较高的催化活性,含各种供电基团和吸电基团的2-氯甲基苯并咪唑和N-4-甲苯磺酰基-2-氨基碘苯衍生物均能较好反应生成相应的咪唑并[1,2-a]喹喔啉,取代基的电子效应对反应有影响,供电子取代基的活性略大于吸电子取代基化合物(图2)。

图2 咪唑并[1,2-a]喹喔啉化合物的合成Fig 2 Synthesis of quinoxaline compounds
2.2结果表征数据 化合物结构鉴定与文献[11]相比,核磁和质谱数据基本一致。
苯并咪唑[1,2-a]喹喔啉(a):白色固体,m. p. 179~180 ℃,产率88%;1H NMR (400 MHz, CDCl3): δ 9.32 (s,1H), 8.54(dd,J= 8.0 Hz, 1.0 Hz, 1H), 8.42 (m, 1H),8.24 (dd,J= 8.0, 1.4 Hz, 1H), 8.13~8.21 (m, 1H),7.77~7.84 (m, 1H), 7.61~7.81 (m,3H);13C NMR (100 MHz, CDCl3): δ 114.4,115.0,122.1,125.0,125.9,125.8,129.9,130.1,131.2,135.9,141.1,144.2,146.0;MS (EI,m/z):220[M+]。
2-甲基苯并咪唑[1,2-a]喹喔啉(b):白色固体,m.p. 167~168 ℃,产率90%;1H NMR(400 MHz,CDCl3):δ 9.21(s,1H),8.38~8.42(m,1H),8.27(s,1H),8.10~8.16(m,1H),8.06(d,J=8.0 Hz,1H),7.59~7.66(m,2H),7.42(dd,J=8.0 Hz,1.2 Hz,1H),2.69(s,3H);13C NMR(100 MHz,CDCl3):δ 22.6,115.4,114.6,122.6,124.6,125.5,126.9,129.6,130.2,131.9,133.9,140.8,141.4,144.0,145.0;MS(EI,m/z):234[M+]。
2-氯苯并咪唑[1,2-a]喹喔啉(c):白色固体,m. p. 177~178 ℃,产率 80%;1H NMR (400 MHz, CDCl3): δ 9.27 (s, 1H), 8.49 (d,J=2.0 Hz, 1H), 8.36~8.40 (m,1H),8.13~8.19 (m, 2H), 7.60 (dd,J= 8.6, 2.1 Hz,IH);13C NMR (100 MHz, CDCl3): δ 114.1, 115.0,122.3, 125.3, 126.0, 126.1, 129.8, 130.2, 132.2, 134.3, 135.8, 140.9, 144.3, 146.2; MS (EI, m/z): 254 [M+]。
9-甲基苯并咪唑[1,2-a]喹喔啉(d):白色固体,m. p. 171~172 ℃,产率 87%;1H NMR (400 MHz, CDCl3): δ 9.25 (q,J= 1.6 Hz,2H), 8.50 (t,J= 9Hz, 2H), 8.30 (d,J= 8.6 Hz, 1H), 8.20 (d,J= 7.8 Hz, 3H),8.03 (d,J= 8.4 Hz, IH),7.92 (s, 1H), 7.78 (m, 2H), 7.62 (t,J= 8.0 Hz, 2H), 7.47 (t,J= 7.6 Hz, 2H), 2.70 (s, 2.4H), 2.63 (s,3.6H);13C NMR (100 MHz, CDCl3): δ 21.6, 22.4, 113.9,114.1,114.8, 114.8,121.5, 121.7, 125.5, 126.5, 127.9, 128.3, 129.7, 129.9,131.1,135.4,135.7,135.9, 135.9, 141.0,141.4, 142.7, 144.8,146.1,146.2; MS (EI, m/z): 234 [M+]。
9-氯苯并咪唑[1,2-a]喹喔啉(e):黄色固体,m. p. 174~176 ℃,产率85%;1H NMR(400 MHz,CDCl3): δ 9.28 (d,J= 3.2 Hz, 1H), 8.33~8.46 (m, 2H), 8.24 (dd,J= 7.8, 1.2 Hz, IH), 8.66~8.13(m, 1H), 7.79~7.86 (m, IH), 7.55~7.69 (m, 2H);13C NMR (100 MHz, CDCl3): δ 114.4, 114.9, 115.1, 121.7, 122.8, 124.3, 125.6, 126.4, 126.5, 126.8, 128.4, 128.9, 130.7, 130.7, 131.4, 131.5, 131.7, 136.9, 138.8, 142.1, 145.2, 145.9, 146.4; MS (EI, m/z): 254 [M+]。
2.3对细胞迁移活性的抑制 咪唑[1, 2-a]喹喔啉结构中当苯环上不含取代基a或者喹喔啉环上含甲基b时,对MCF-7细胞具有的抑制作用不强(c>40 μmol/L);当化合物喹喔啉环2位上是引入取代的Cl原子c时,对MCF-7的抑制浓度达到20 μmol/L;而当喹喔啉环9位上取代是Cl原子e时,抑制细胞生长浓度均>20 μmol/L。由此可见, 咪唑[1, 2-a]喹喔啉结构中含有吸电子的Cl时, 化合物针对抑制MCF-7细胞生长具有较高活性,最低抑制浓度为20 μmol/L,细胞毒活性明显增大。
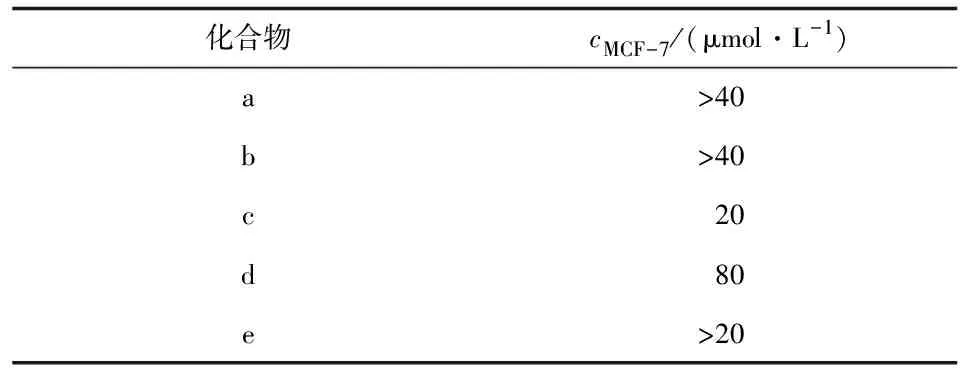
表1 咪唑并[1,2-a]喹喔啉对 MCF-7细胞迁移活性的抑制
2.4化合物对乳腺癌细胞MCF-7和SKBR3增殖的抑制作用 MCF-7和 HER2阳性的SKBR3细胞经不同浓度的新化合物处理48 h后,MTT法检测咪唑[1,2-a]喹喔啉化合物对乳腺癌不同种类细胞的增殖和抑制作用。结果表明:随着药物浓度的增加,喹喔啉化合物抑制MCF-7和SKBR3细胞增殖的能力逐步增强,针对MCF-7和SKBR3的半数抑制浓度IC50见表2。其中化合物c和e具有较强的抑制乳腺癌细胞增殖的作用,IC50均在12~18 μmol/L,经过对比实验,该类化合物的半抑制率浓度略差于紫杉醇活性,具备进一步通过结构改造,筛选出具备高抗肿瘤活性的喹唑啉化合物的潜力,并为进一步进行抗肿瘤活性的体内研究提供潜在药物分子。
2.5统计结果 经过One Way ANOWA分析统计,在MCF-7细胞组中,化合物c和e分别与化合物a的IC50比较,差别具有统计学意义(P<0.05)。

表2 咪唑[1,2-a]喹喔啉对乳腺癌细胞的抑制作用
IC50:半数抑制浓度.
3 讨 论
通过微波辐射法,采用镍催化剂高效合成了5个咪唑并[1,2-a]喹喔啉衍生物。含吸电子基团的化合物其细胞毒活性明显大于含供电子基团的化合物,尤其含Cl原子取代的化合物。在针对MCF-7和SKBR3肿瘤细胞抑制作用实验中,2-氯苯并咪唑[1,2-a]喹喔啉(c)和9-氯苯并咪唑[1,2-a]喹喔啉(e)也对MCF-7和SKBR3肿瘤细胞增殖抑制活性好。含有Cl原子的苯并咪唑[1,2-a]喹喔啉活性相对其他化合物强,与其他化合物相比均有显著差异性(P<0.05)。由此可见,含有Cl原子的苯并咪唑[1,2-a]喹喔啉是具有高度活性的先导化合物,可进一步通过构效关系对其进行结构改造和靶点优化,筛选出活性更好的新型抗肿瘤药物。
