Mo-Mn/TiO2催化剂的协同脱硝脱汞特性
2018-03-02段钰锋陈亚南丁卫科李春峰王双群东南大学能源与环境学院能源热转换及其过程测控教育部重点实验室江苏南京210096
胡 鹏,段钰锋,陈亚南,周 强,朱 纯,丁卫科,李春峰,刘 猛,王双群 (东南大学,能源与环境学院,能源热转换及其过程测控教育部重点实验室,江苏 南京 210096)
目前,选择性催化还原法(SCR)[1-2]是国际上工业应用最广泛、最成熟的烟气脱硝技术.据文献报道,SCR脱硝装置不仅可以有效控制NOx的排放[3],也能将含量高且难以去除的元素汞(Hg0)氧化成易溶于水的化合态汞(Hg2+)[4-5],再经后续的湿法烟气脱硫装置(WFGD)去除[6-7].因此,可利用燃煤电站的现有脱硝设备对汞进行协同脱除,在不降低脱硝效率的同时尽可能提高催化剂对汞的去除效率.这样不仅符合我国当前的国情,也大大提高了SCR脱硝装置的经济性,具有重要的学术意义和工业前景.国内外已商业化的SCR脱硝催化剂(V2O5-WO3/MoO3-TiO2)活性温度窗口为300~400℃,SCR装置布置在除尘器之前时,催化剂易受飞灰等的影响出现磨损、堵塞和烧结等现象导致其寿命降低,且催化剂表面的活性成分可将SO2氧化形成SO3,进而形成硫酸盐腐蚀后续设备[8].另外,此温度区间极大的偏离了烟气中汞去除的活性温度窗口,对汞的脱除效率不高[9].因此,SCR脱硝协同脱汞技术的核心是高效经济的低温(<300℃)SCR催化剂的开发.
TiO2的基本存在形态为含有大量Lewis酸性位的锐钛矿型[10],此晶体结构具有较高的SCR活性且硫酸盐在其表面不易沉积,因此TiO2作为低温SCR催化剂的载体而被广泛研究[11-12].锰元素具备多种不稳定价态且相互转化所需的活化能较低[13-14],因此具有较强的低温氧化还原活性,使其成为低温SCR催化剂领域研究的热点[15-20]. 200℃时MnOx/TiO2催化剂可实现对NOx和Hg0的高效脱除[16],催化剂表面的化学吸附氧和晶格氧、Mn4+/Mn3+等极大的影响着催化剂的活性[18].大量研究表明[9,20-21,29],采用Mo对锰氧化物催化剂进行改性,可同时提高其催化效果和抗硫性能.此外,MoOx可以同时提供有利于NH3吸附的Lewis酸性位和有助于Hg0脱除的Bronsted酸性位[20],在SCR催化剂中掺杂Mo可进一步提高其催化氧化性能,且Mo的促进效果在与Mn结合使用时更加显著[21].综上所述,掺杂Mn或Mo的SCR催化剂可克服传统SCR催化剂的部分局限性,然而,将二者的性能结合用于NO和Hg0协同脱除的报道却不多见.
本文采用浸渍法制备了Mo-Mn/TiO2催化剂,考察反应温度、HCl和SO2对其模拟烟气协同脱硝脱汞的影响,并通过对SO2中毒反应前后的催化剂进行XRD、H2-TPR、XPS等表征,以期获得催化剂的脱硝脱汞机制及抗硫中毒机理.
1 实验方法
1.1 催化剂的制备
本文采用浸渍法制备Mo-Mn/TiO2催化剂,具体过程如下:首先取50mL超纯水放入小烧杯中,将一定体积的Mn(NO3)2溶液(国药集团化学试剂有限公司,质量分数为50%,分析纯)和相应质量的(NH4)6Mo7O24·4H2O试剂(国药集团化学试剂有限公司,分析纯)溶于上述小烧杯,并在常温下磁力搅拌20min,再称取10g TiO2(Degussa P25)粉末到以上溶液,继续搅拌2h使其充分混合,将所得混合溶液置于105℃的烘箱中烘干后在空气气氛下450 ℃煅烧4h,研磨、筛分至60~80目,存放于干燥皿中备用.单次实验催化剂用量为2.5g.
1.2 催化剂表征
反应前后催化剂的表面晶体结构采用德国BRUKER公司的D8ADVANCE X射线衍射仪测定;氧化还原性能采用日本Microtrac BEL公司的BELCAT-M自动化程序升温化学吸附仪测定;表面元素状况采用美国Thermo Fisher Scientific公司的ESCALAB250Xi型X射线光电子能谱分析仪测定,并使用XPSPeak41分峰软件对催化剂表面各原子光谱进行分析.
1.3 固定床实验装置
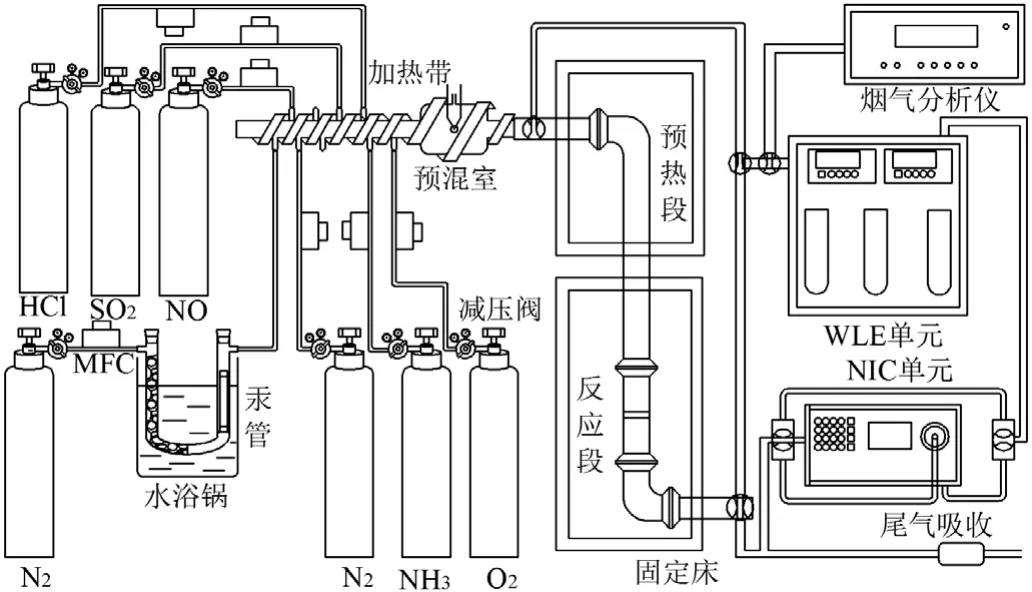
图1 固定床实验系统示意Fig.1 Schematic diagram of fixed bed system
催化剂的脱硝脱汞性能测试在实验室固定床装置上进行,如图1所示.管式炉呈两段式(程序升温控制仪精确控温),分别为模拟气体预热段和气固催化反应段.反应管垂直放置,内径为15mm,中间填置砂芯作为催化剂的支撑床层.模拟烟气组成为0.050% NO、0.050% NH3、5%O2、(60±0.5) µg/m3Hg0、平衡N2、载汞N2,气体总流量为1.0L/min,各路气体流量由质量流量计(北京七星华创电子股份有限公司)精确控制.催化剂用量为2.5mL,空速为24000h-1,反应温度区间为120~280℃.反应进出口NO浓度用Seitron C500烟气分析仪(意大利)进行在线检测,反应前后Hg0浓度采用EMP-2/WLE-8在线测汞仪(日本株式会社)进行实时监测.实验过程中为避免气流的扰动对实验结果造成影响,催化剂活性测试均在反应稳定30min后进行.
1.4 催化剂活性评价方式
依据反应前后汞蒸气及NO气体浓度变化来评价催化剂的脱硝协同脱汞性能.
Hg0的脱除效率η(%)为:

式中:Cin和Cout分别表示固定床实验装置进出口Hg0的质量浓度, µg/m3.
NO的脱除效率β(%)为:
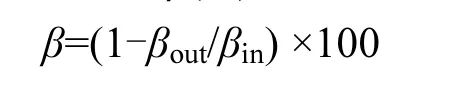
式中:βin和βout分别表示固定床实验装置进出口NO的质量浓度, mg/m3.
2 结果与讨论
2.1 反应温度的影响
图2为反应温度对催化剂Mo-Mn/TiO2协同脱硝脱汞性能的影响.
在所考察的温度段内,Mo-Mn/TiO2对Hg0的脱除率随反应温度的提高表现出先略微上升后下降的趋势.在120℃时,催化剂的脱汞效率为96.4%,而当温度提高至160℃,其脱汞效率上升至最高的98.5%.继续升高反应温度,其脱汞效率反而逐渐降低,当温度达到280℃时,Mo-Mn/ TiO2对Hg0的脱除效率已降至75.4%.燃煤烟气中汞的脱除是物理吸附和化学氧化共同作用的结果,当反应温度较低时,难以满足脱汞反应中化学氧化所需的能量,此时物理吸附起主导作用;随着反应温度的逐步上升,吸附在催化剂表面的Hg0会发生脱附,致使物理吸附减弱,但反应温度的升高,将提供化学氧化所需能量且加快反应速率,此时化学氧化对脱汞过程起到关键作用[22].由此可知,Mo-Mn/TiO2对Hg0的脱除效率随温度的增加先上升,此阶段遵循化学氧化规律,而其脱汞效率在160℃以后的降低,说明物理吸附对脱汞反应同样具有重要作用.

图2 反应温度对协同脱硝脱汞活性的影响Fig.2 Effect of reaction temperature on denitration and mercury removal
随着反应温度的升高,催化剂Mo-Mn/TiO2对模拟烟气中NO的脱除效果却呈现出不一样的趋势.在120℃时,Mo-Mn/TiO2的脱硝效率仅为19.2%,当反应温度升至200℃,则可获得91.8%的脱硝效率,继续升高反应温度,Mo-Mn/TiO2可将NO完全转化.当反应温度较低时,NH3-SCR过程中的NO2与NH3反应生成NH4NO3会堵塞催化剂的孔道,导致催化剂活性的降低,是低温下催化剂脱硝效率不高的主要原因[23].
在本文所研究温度区间内,Mo-Mn/TiO2对NO的脱除效率随着温度的升高而增加,表明此脱硝过程受化学反应步骤控制[24].综上所述,催化剂Mo-Mn/TiO2的协同脱硝脱汞的最佳温度点为200℃,此温度下的脱汞效率达到88.9%,而NO的脱除率亦可达91.8%.
2.2 HCl的影响
催化剂单独作用于Hg0的脱除效率很低,而元素O和Cl是SCR脱硝催化剂催化氧化Hg0转化为Hg2+的主要推动力.图3为反应温度为200℃时的HCl浓度对催化剂Mo-Mn/TiO2协同脱硝脱汞性能的影响.
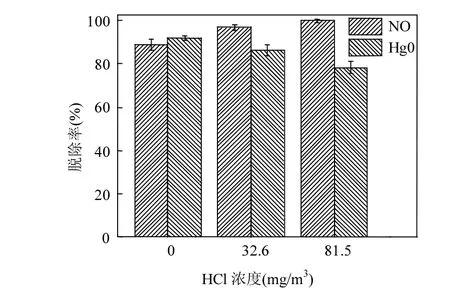
图3 HCl浓度对协同脱硝脱汞活性的影响Fig.3 Effect of HCl concentration on denitration and mercury removal
结果表明,当模拟烟气中存在O2时,适当增加烟气中HCl的浓度便可显著提高催化剂的脱汞效率.当向烟气中添加32.6mg/m3HCl时,Mo-Mn/TiO2的脱汞效率由88.85%升至97.4%,说明HCl可大大促进催化剂对Hg0的去除.这是由于气相HCl会吸附在催化剂表面形成吸附态的氯氧化物活性位,然后在含O2气氛下与Hg0发生反应形成中间产物HgCl,最后再与吸附态的HCl反应被氧化为HgCl2从催化剂表面脱附出来[25].
然而,促进Hg0被氧化的过程恰恰阻碍了催化剂对NO的脱除.当HCl浓度为32.6mg/m3和81.5mg/m3时,Mo-Mn/TiO2的脱硝效率从91.8%分别下降至86.2%和78.4%.首先,HCl可与NH3竞争吸附催化剂表面的活性位点,影响NH3的吸附数量,抑制脱硝反应的进行;其次,吸附态HCl会与NH3反应生成铵盐,降低烟气中还原剂NH3的浓度,导致NH3-SCR反应不完全.因此,向模拟烟气中加入HCl将导致脱硝效率的持续下降.反应过程如式(1)~(3)所示:
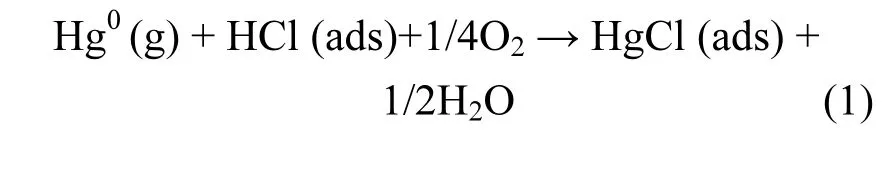
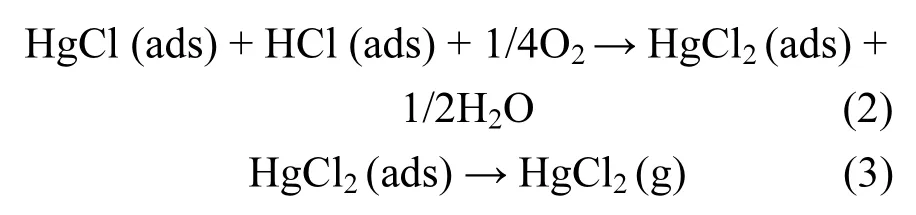
2.3 SO2的影响
2.3.1 SO2浓度的影响 图4为反应温度为200℃时SO2浓度对催化剂Mo-Mn/TiO2协同脱硝脱汞效率的影响.
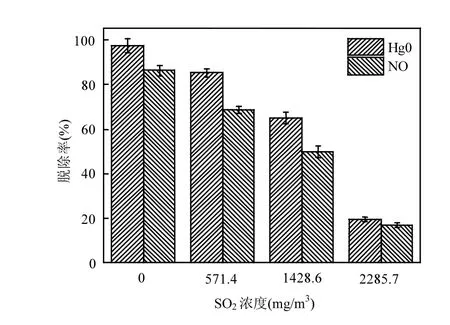
图4 SO2浓度对协同脱硝脱汞活性的影响Fig.4 Effect of SO2 concentration on denitration and mercury removal
当模拟烟气中不存在SO2时, Mo-Mn/TiO2的脱汞效率高达97.4%(含32.6mg/ m3HCl).向气氛中加入571.4mg/m3SO2,催化剂对Hg0的脱除活性呈现出一定程度的减弱,特别是当SO2浓度增加至2285.7mg/m3,其脱汞效率明显下降至27.6%.表明SO2对Hg0的氧化去除产生强烈抑制作用,且SO2浓度越高,抑制作用越明显.
原因可能在于:(1)烟气中的SO2与HCl在催化剂表面形成竞争吸附,HCl向催化剂活性位点的扩散速率降低,导致催化剂的脱汞效果变差[26]; (2)HCl和SO2与NH3反应生成铵盐,覆盖催化剂表面的活性位点及堵塞催化剂孔道,且在此200℃反应温度下,铵盐无法分解,因此致使催化剂的活性下降.
与催化剂的脱汞活性相比,SO2对Mo-Mn/TiO2的脱硝性能展现出相同的作用结果.模拟烟气中无SO2时,催化剂的脱硝效率为86.2%,当烟气中存在一定量的SO2时,催化剂对NO的去除效果显著减弱,如向烟气中添加571.4mg/m3SO2,催化剂的脱硝效率下降至68.5%,当SO2浓度继续增至2285.7mg/m3,脱硝效率仅为16.9%.与脱汞过程相比,催化剂的脱硝效率下降的更为迅速,可能因为:(1)SO2和HCl同时与NH3竞争催化剂表面的活性位点,致使NH3无法充分被吸附,导致脱硝效率明显下降;(2)形成的卤化铵盐和硫酸铵盐难以分解,堵塞催化剂孔道,使NO无法高效脱除.综上所述,SO2对Mo-Mn/TiO2的脱硝脱汞过程均起到阻碍作用.
2.3.2 SO2对催化剂理化特性的影响
(1) XRD分析 图5为催化剂Mo-Mn/TiO2协同脱硝脱汞研究中SO2中毒反应前后的XRD图谱.
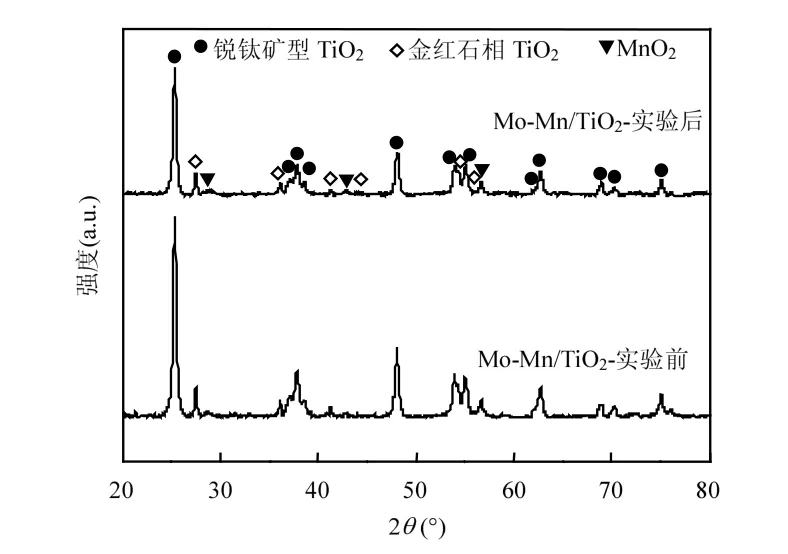
图5 反应前后催化剂的XRD图谱Fig.5 XRD spectra of fresh and used catalysts
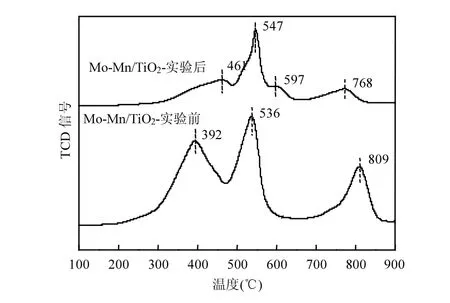
图6 反应前后催化剂的H2-TPR图谱Fig.6 H2-TPR spectra of fresh and used catalysts
结果表明,催化剂中的Ti均主要以锐钛矿型TiO2存在;此外,在催化剂表面可明显观察到微弱的MnO2特征衍射峰,说明MnO2在TiO2载体上分散性较好且Mn4+为催化剂的主要活性价态.对比SO2中毒前后的Mo-Mn/TiO2表征结果可知,被SO2毒化之后,催化剂的晶体类型无显著变化,可推断出SO2对催化剂的基本物质成分不产生影响.另外,没有观察到新晶型的出现,且原有各物质成分衍射峰的峰值明显降低,表明催化剂与SO2反应后形成的硫物种含量较少或均以无定形状态覆盖于催化剂表面,因此无法被明显检测到.
(2) H2-TPR分析 图6为催化剂Mo-Mn/TiO2协同脱硝脱汞研究中SO2中毒反应前后的H2-TPR图谱.
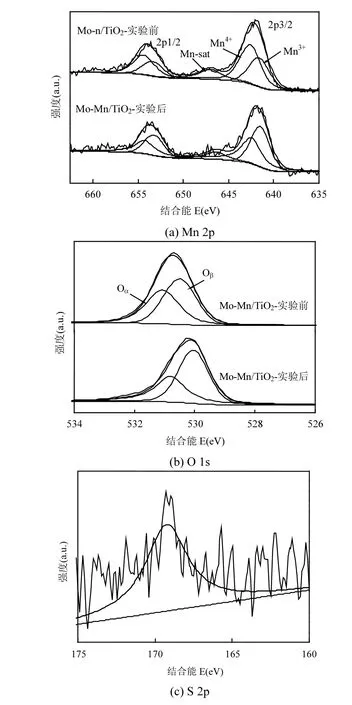
图7 反应前后催化剂的XPS图谱Fig.7 XPS spectra of fresh and used catalysts
研究表明,锰氧化物的还原一般具有两种途径,分别为MnO2→ Mn2O3/Mn3O4→ MnO[27]或MnO2→ Mn2O3→ Mn3O4→ MnO[28],MoO3的还原过程通常为Mo6+→ Mo4+→ Mo (400~800℃)[29].
表征结果显示,反应前后的两种样品均存在3个特征还原峰,第一个峰应归属于MnO2→Mn2O3的还原,第二个峰为Mn2O3和MoO3的还原,最后一个峰为Mo-Mn复合氧化物的还原.相比之下,Mo-Mn/TiO2与SO2反应后,位于约392℃和809℃的还原峰基本消失,原因可能是反应生成的硫酸盐覆盖于催化剂表面,阻碍了锰和钼氧化物的氧化还原;此外,催化剂在597℃出现了新的还原峰,应为Mo6+被还原成Mo4+的过程,Mo元素的添加一定程度上抑制了活性组分Mn的硫酸化,对催化剂起保护作用,此峰可能是硫酸钼的特征峰.
(3) XPS分析 催化剂Mo-Mn/TiO2协同脱硝脱汞研究中SO2中毒反应前后的XPS图谱及表面元素含量变化分别列于图7和表1.
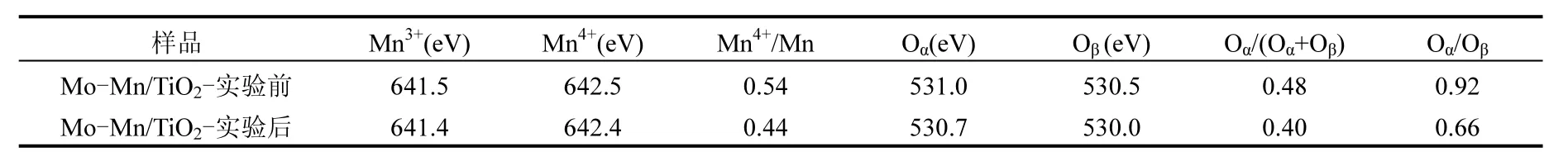
表1 反应前后催化剂表面各元素浓度Table 1 Concentration of element on the surface of fresh and used catalysts
从Mn 2p表征结果可知,Mn 2p3/2和Mn 2p1/2的特征峰分别位于642eV和654eV左右,且锰物种是以Mn4+、Mn3+和Mn-sat的混合态存在于催化剂中.由表1中催化剂中毒反应前后Mn元素价态变化可知,中毒后Mn4+的百分含量明显降低,这是因为中毒后催化剂表面的锰被硫酸化,Mn4+失去氧导致其价态降低且难以恢复,因此无法完成氧化还原循环.
但从H2-TPR分析结果可知催化剂表面生成了硫酸钼,可以推断出Mo元素可以保护部分活性组分Mn不被硫酸化.而对于O 1s的表征结果,催化剂Mo-Mn/TiO2的晶格氧Oβ(O2-)和表面吸附氧Oα(或O-)的特征峰分别位于约530.5eV和531eV处,而Oα更能促进催化氧化反应过程的进行[30].对比硫中毒前后的两种样品,催化剂Mo-Mn/TiO2-实验后的表面吸附氧Oα含量降低,说明反应过程中SO2的吸附和氧化消耗了催化剂表面的活性氧,而晶格氧Oβ含量增加,这与中毒后催化剂表面覆盖的和关系巨大,说明硫酸盐或硝酸盐等物质覆盖于催化剂表面,致使催化剂活性降低.从S 2p能谱来看,XPSPEAK41分析证实为.
2.3.3 SO2对催化剂的毒化机理 表2为几种汞标准化合物的热脱附(TPD)结果.
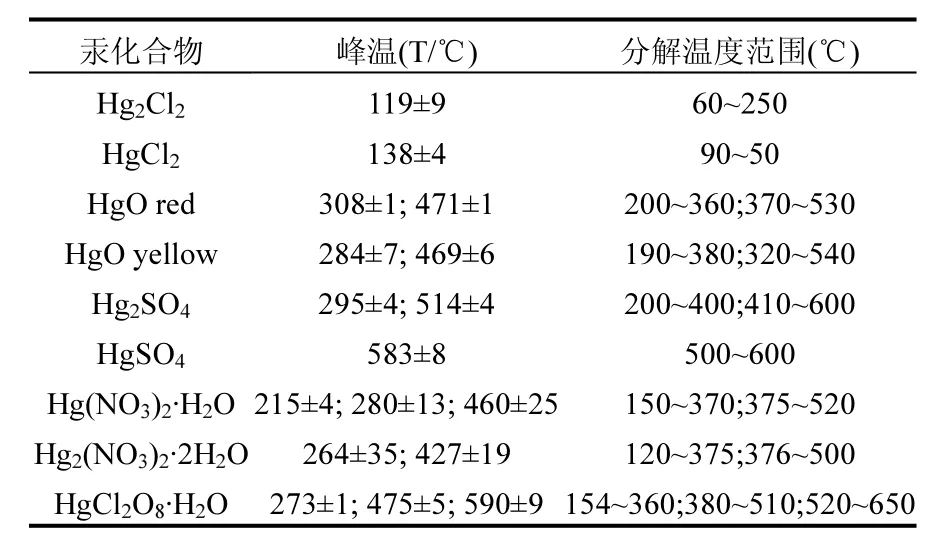
表2 汞标准化合物热脱附结果[32]Table 2 Thermal desorption of mercury standard compounds
当升温速率相同时,不同的汞化合物会在其特定的某一温度段内发生分解,因此可通过TPD曲线分析获得催化剂表面存在的汞形态.Wang[33]研究得知实验条件的不同将导致汞化合物分解峰值所对应的温度相异,但差别不大.在SCR+Hg0和SCR+Hg0+ SO2气氛中反应后的Mo-Mn/TiO2催化剂的TPD曲线如图8所示.
实验结果显示,在含有SO2气氛中反应后的催化剂表面吸附的汞化合物量更多,表明SO2阻碍汞的催化氧化过程.并且TPD曲线具有两个峰,推断有新物质的生成.对比表2和图8中结果表明反应后的催化剂表面无HgSO4的存在,但可能生成了Hg2SO4.气相Hg0吸附在催化剂表面形成吸附态Hg0,与催化剂表面被吸附氧化生成的SO3反应生成Hg2SO4,同时Mn4+被还原为Mn3+,如式(4)和(5)所示.Hg2SO4在反应温度200℃时无法分解而沉积于催化剂表面.

图8 Mo-Mn/TiO2催化剂的汞脱附曲线Fig.8 The mercury desorption curve of Mo-Mn/TiO2 catalyst
此外,图7(c)分析得知反应后催化剂表面的硫物种是以SO42-形态存在的,因此可知覆盖于催化剂表面造成其失活的硫物种主要是金属氧化物被硫酸化生成的硫酸盐及SO2与NH3反应形成硫酸铵盐和上述的Hg2SO4等,这些含硫物质在催化剂表面不断沉积且难以分解,导致催化剂孔道的堵塞,造成催化剂的钝化,如式(4)~式(7)[34].
根据图7(a)和(b)分析发现,活性组分Mn4+和Oα含量的显著降低,是使得催化剂脱硝脱汞性能下降的另一主要原因.另外,SO2的浓度远远大于Hg0的通入量,严重影响到Hg0的吸附,抑制催化剂的脱汞过程;SO2也会与NH3竞争催化剂表面的活性位点,从而不利于脱硝反应的进行.
2.4 脱硝脱汞机理分析
汞的脱除过程主要分为物理吸附和化学氧化.图9为Mo-Mn/TiO2催化剂在SCR+Hg0和SCR+Hg0+HCl气氛下的汞穿透率曲线及其TPD曲线.根据origin卷积公式得出催化剂脱附汞量分别占进口汞总量的4%和1%,可得知Mo-Mn/ TiO2对Hg0的脱除以催化氧化为主.
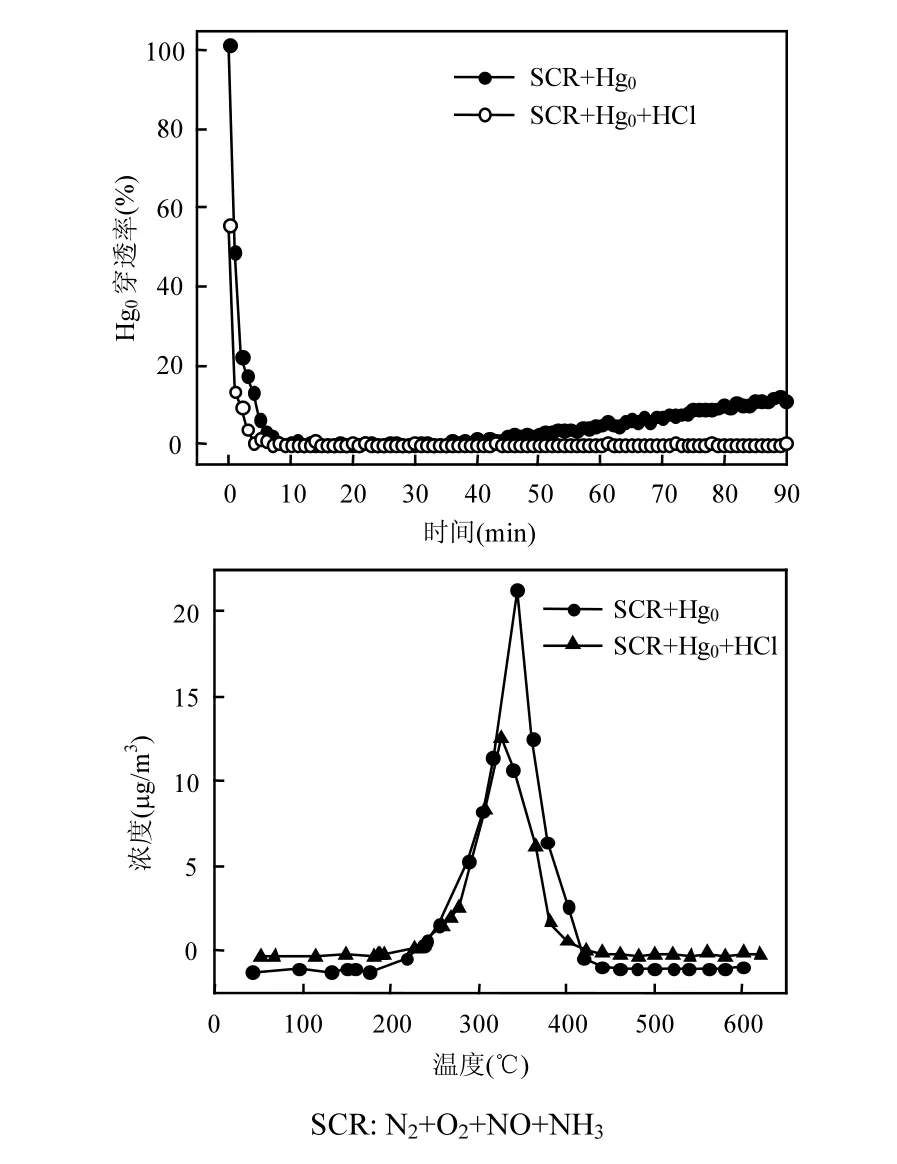
图9 Mo-Mn/TiO2催化剂的汞穿透率曲线和汞脱附曲线Fig.9 The mercury breakthrough curve and desorptioncurve of Mo-Mn/TiO2 catalyst
由图9和表2可知,催化剂通过催化氧化Hg0转化为HgO而实现脱汞过程.一方面,烟气中的Hg0首先吸附在催化剂表面的活性位点,形成吸附态Hg0(ads);Hg0(ads)再与催化剂活性组分MnOx和MoOx中的晶格氧Oβ反应生成HgO (ads),而此时金属氧化物产生缺陷位,氧气与具有缺陷位的金属氧化物反应并使其恢复晶体结构,继续参与到循环过程中;最后,吸附态HgO (ads)从催化剂表面脱附向气相迁移形成HgO (g).可以归纳为[35-36]:
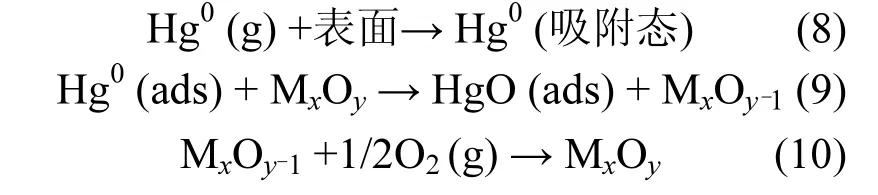
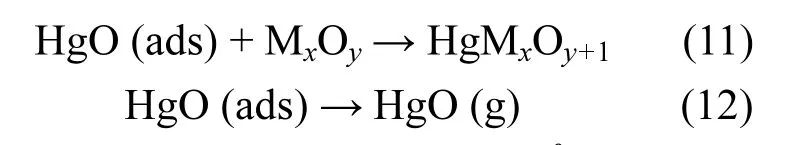
另一方面,在SCR的气氛中,Hg0也许被氧化形成了Hg(NO3)2.尽管NH3-SCR脱硝反应的还原剂NH3会和Hg0的吸附产生竞争,但SCR的烟气成分NO极易被催化剂氧化为NO2,NO2具有很强的氧化能力,反而对Hg0的脱除过程起到促进作用,Hg0转化为Hg(NO3)2,但在200℃的反应温度下,Hg(NO3)2分解为HgO,如式(13)~(16)所示[37-38].另外,催化剂表面活性组分众多的酸性位点和化学吸附氧,可缓解NH3带来的抑制作用.因此,Mo-Mn/TiO2展现出较为理想的脱硝协同脱汞性能.从图9中的结果可以看出,HCl促进催化剂对Hg0的脱除,但在HCl气氛中反应后的催化剂脱附峰的峰值明显低于无HCl存在时反应后催化剂的脱附峰值,可推断HCl更加促进Mo-Mn/TiO2的催化氧化过程.HCl的加入,使得峰温向低移动,表明有其它汞化合物生成,但此汞化合物极易挥发,随尾气逸出.
Mo-Mn/TiO2催化剂表面的SCR脱硝机理:首先气态中的NO被催化剂表面MnO2中的晶格氧所氧化生成NO2,同时高价态的Mn4+被还原为Mn3+,另一个途径是NO可以通过与催化剂中的Mn3+和Mn4+共同反应生成NO2,而Mn3+和Mn4+均被还原成更低价态的锰氧化物;与此同时,气相中的还原剂NH3吸附在催化剂表面的活性位点,可以直接与弱吸附态的NO或被氧化生成的NO2反应,生成N2和H2O,如式(13)、(14)、(17)和(18)所示[19,38].
整个协同脱硝脱汞反应过程中,活性更好的Mn4+被还原为低价态的锰氧化物无法继续参与反应.但催化剂中的助活性组分MoO3能起到将低价态的锰氧化物转化为性能好的Mn4+的作用,使活性组分MnO2再次参与到脱硝反应中.而因反应被还原成的MoO2可依靠O2的持续补充再次形成MoO3.因此,可使脱硝反应循环往复,如式(19)~(21)[38].
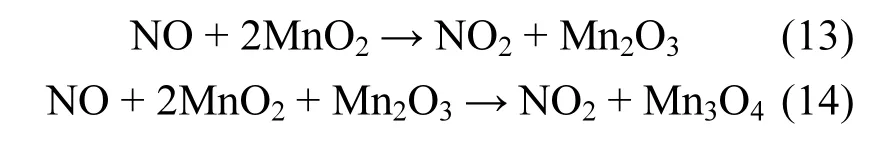
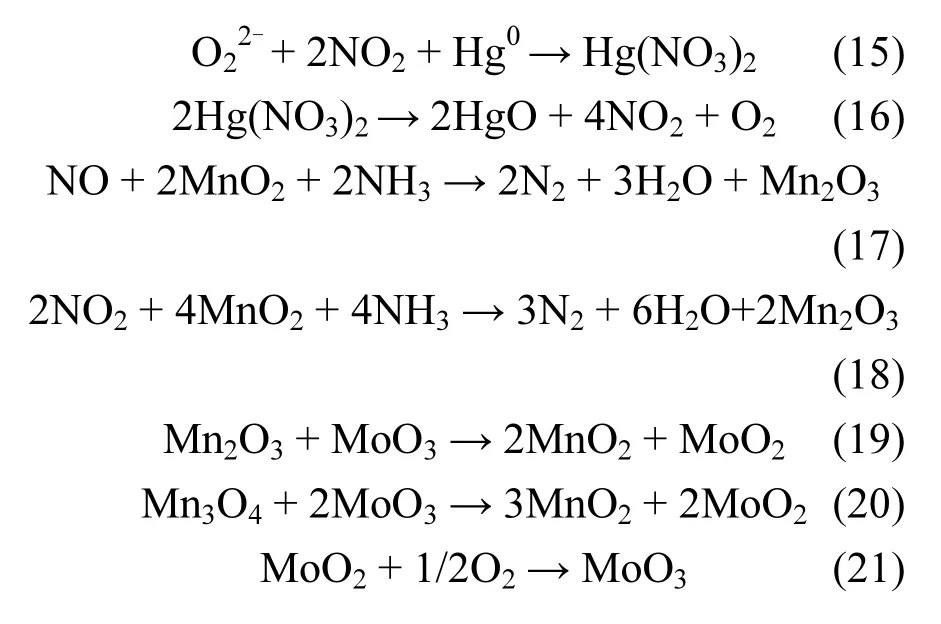
3 结论
3.1 催化剂Mo-Mn/TiO2展现出了良好的协同脱硝脱汞性能.在200℃时的Mo-Mn/TiO2可兼具最好的脱硝和脱汞效率.
3.2 模拟烟气中HCl的添加有利于Mo-Mn/TiO2对汞的氧化脱除,却会阻碍脱硝反应的进行;SO2对催化剂的脱硝和脱汞过程均起到抑制作用,但硫酸盐的积累和活性成分的消耗是致使催化剂失活的主要原因.
3.3 氧元素对催化剂的脱硝脱汞过程均起到重要作用;Mo-Mn/TiO2催化剂的脱汞过程以催化氧化为主,金属氧化物中的晶格氧可将Hg0氧化为HgO,从而实现汞的去除.
[1] 胡 强,熊志波,白 鹏,等.铈钛掺杂促进铁氧化物低温SCR脱硝性能的机理 [J]. 中国环境科学, 2016,36(8):2304-2310.
[2] 熊志波,金 晶,路春美,等.铁基催化剂的微波水热处理对其SCR脱硝性能的影响 [J]. 中国环境科学, 2014,34(7):1785-1789.
[3] 董晓真.同时脱硝脱汞催化剂研究进展 [J]. 山东化工,2016,45(19):58-59.
[4] 邢微波,路 好,张安超,等.Ag/AgCl可见光催化剂湿法脱除烟气中单质汞 [J]. 中国环境科学, 2017,37(2):503-510.
[5] 张安超,张洪良,宋 军,等.Mn-Co/MCM-41吸附剂表征及脱除烟气中单质汞研究 [J]. 中国环境科学, 2015,35(5):1319-1327.
[6] Yamaguchi A, Akiho H, Ito S. Mercury oxidation by copper oxides in combustion flue gases [J]. Powder Technology, 2008,180(1):222-226.
[7] 高 鹏,向 军,张安超.壳聚糖吸附剂脱除燃煤模拟烟气中汞的试验研究 [J]. 中国环境科学, 2010,30(6):733-737.
[8] 廖伟平.Mn-Ce/TiO2催化剂低温选择性催化还原NO机理研究 [D]. 南京:南京师范大学, 2012.
[9] 王晓刚,张益坤,丁 峰,等.SCR催化剂对汞的催化氧化研究进展 [J]. 环境科学与技术, 2014,37(4):68-73.
[10] 姚 杰,仲兆平.蜂窝状SCR脱硝催化剂成型配方选择 [J].中国环境科学, 2013,33(12):2148-2156.
[11] Pena D A, Uphade B S, Smirniotis P G. TiO2-supported metal oxide catalysts for low-temperature selective catalytic reduction of NO with NH3: I. Evaluation and characterization of first row transition metals [J]. Journal of Catalysis, 2004,221(2):421-431.
[12] 石研妮.多级大孔-介孔Mn/TiO2低温SCR脱硝催化剂的制备及其反应机理研究 [D]. 大连:大连理工大学, 2014.
[13] 陈 玲,李彩亭,高 招,等.MnOx/HZSM-5去除烟气中元素态汞的实验研究 [J]. 中国环境科学, 2010,30(8):1026-1031.
[14] 唐晓龙,郝吉明,易红宏,等.活性炭改性整体催化剂上低温选择性还原NOx[J]. 中国环境科学, 2007,27(6):845-850.
[15] 郑玉婴,汪 谢.Mn基低温SCR脱硝催化剂的研究进展 [J].功能材料, 2014,11(45):11008-11012.
[16] Ji L, Pavani M S, Panagiotis G S, et al. Manganese Oxide/Titania materials for removal of NOxand elemental mercury from flue gas [J]. Energy & Fuels, 2008,22(4):2299-2306.
[17] He J, Gunugunuri K R, et al. Simultaneous Removal of Elemental Mercury and NO from Flue Gas Using CeO2Modified MnOx/TiO2Materials [J]. Energy & Fuels. 2013,27:4832-4839.
[18] Wang Y Y, Shen B X, et al. Simultaneous Removal of NO and Hg0from Flue Gas over Mn-Ce/Ti-PILCs [J]. Environmental Science & Technology, 2015,49:9355-9363.
[19] 王晓梅.W-Mn-TiO2复合金属氧化物的低温NH3-SCR性能的研究 [D]. 大连:大连理工大学, 2015.
[20] 闫智锋.Mo/HZSM-5催化剂上NH3选择性催化还原NO反应机理的密度泛函理论研究 [D]. 太原:太原理工大学, 2015.
[21] 陈 杰,晏乃强,瞿 赞,等.强化SCR脱硝催化剂转化零价汞的初步研究 [J]. 环境科学与技术, 2013,36(5):86-88.
[22] Yan R, Liang D T, Tsen L, et al. Bench-scale experimental evaluation of carbon performance on mercury vapour adsorption [J]. Fuel, 2004,83(17/18):2401-2409.
[23] 陈进生.火电厂烟气脱硝技术-选择性催化还原法 [J]. 北京:中国电力出版社, 2008:99-104.
[24] Lu P, Li C T, Zeng G M, et al. Low temperature selective catalytic reduction of NO by activated carbon fiber loading lanthanum oxide and ceria [J]. Applied Catalysis B:Environmental, 2010,96:157-161.
[25] 李 锋,於承志,张 朋,等.低SO2氧化率脱硝催化剂的开发[J]. 电力科技与环保, 2010,26(4):18-21.
[26] 唐 念,胡将军,盘思伟,等.Mn-Nb复合催化剂脱汞性能及抗硫特性试验研究 [J]. 环境科学与技术, 2015,38(2):184-188.
[27] Xu W, He H, Yu Y. Deactivation of a Ce/TiO2catalyst by SO2in the selective catalytic reduction of NO by NH3[J]. The Journal of Physical Chemistry C, 2009,113:4426-4432.
[28] Jiang B, Deng B, Zhang Z, et al. Effect of Zr addition on the low-temperature SCR activity and SO2tolerance of Fe-Mn/Ti catalysts [J]. The Journal of Physical Chemistry C, 2014,118:14866-14875.
[29] 李泽英,唐 清,左赵宏.载体对V2O5-MoO3/TiO2催化剂表面性质及其选择性催化还原脱硝性能的影响 [J]. 工业催化,2016, 24(7):41-48.
[30] Zhang J X, Zhang S L, Cai W, et al. The characterization of CrCe-doped on TiO2-pillared clay nanocomposites for NO oxidation and the promotion effect of CeOx[J]. Applied Surface Science, 2013,268:535-540.
[31] Romano E J, Schulz K H. A XPS investigation of SO2adsorption on ceria-zirconia mixed-metal oxides [J]. Applied Surface Science, 2005,246(1-3):262-270.
[32] Rumayor M, Díaz-Somoano M, Lopez-Anton M A, et al.Mercury compounds characterization by thermal desorption [J].Talanta, 2013,114:318-322.
[33] Wang F Y, Wang S X, Meng Y, et al. Mechanisms and roles of fly ash composition on the adsorption and oxidation of mercury in flue gas from coal combustion [J]. Fuel, 2016,163:232-239.
[34] 杜学森.钛基SCR脱硝催化剂中毒失活及抗中毒机理的实验和分子模拟研究 [D]. 杭州:浙江大学, 2014.
[35] 樊小鹏,李彩亭,曾光明,等.CuO-CeO2/AC吸附燃煤烟气中元素汞的实验研究 [J]. 环境工程学报ISTIC, 2010,4(2):393-397.
[36] 游华伟.MnOx-CeO2/γ-A12O3催化剂脱硝脱汞的实验研究[D]. 武汉:华中科技大学, 2012.
[37] 王鹏鹰,苏 胜,向 军,等.低温SCR催化剂脱硝脱汞实验研究 [J]. 燃烧科学与技术, 2014,20(5):423-427.
[38] 郭 静,李彩亭,路 培,等.CeO2改性MnOx/Al2O3的低温SCR法脱硝性能及机制研究 [J]. 环境科学, 2011,32(8):2240-2246.
