吸烟COPD模型大鼠肺组织内质网相关凋亡蛋白CHOP的表达*
2018-03-01甘桂香胡瑞成谭双香
甘桂香, 胡瑞成, 谭双香
(湖南省人民医院呼吸内科, 湖南 长沙 410016)
慢性阻塞性肺疾病(chronic obstructive pulmonary disease,COPD)是一种严重危害人类健康的常见病、多发病,其病情常呈进行性发展,最终导致患者劳动能力丧失和死亡,给社会造成沉重的经济负担。香烟烟雾(cigarette smoke, CS)中的氧化性物质可通过内质网应激(endoplasmic reticulum stress,ERS)导致肺结构细胞凋亡,促进COPD的发生发展[1]。CCAAT/增强子结合蛋白同源蛋白(CCAAT/enhancer-binding protein homologous protein,CHOP)在正常生理状态下基本检测不到,但在ERS状态下表达显著升高,是ERS的标志。本研究通过TUNEL法检测肺组织细胞的凋亡,并通过检测PERK/eIF2α信号通路在COPD大鼠中的表达情况,为COPD的防治提供新方向。
材 料 和 方 法
1动物
清洁级雄性Wistar大鼠40只(湖南中医药大学实验动物中心提供),动物合格证编号为SCXK(湘)2013-0003,体重(200±20) g,鼠龄10周。将大鼠随机分为对照(control)组、吸烟2个月(exposed to CS for 2 months, CS-2)组、吸烟4个月(CS-4)组及戒烟1个月(ex-smoking, Ex-S)组,每组10只。参照王慧等[2]方法建立COPD大鼠模型,每日将大鼠置于自制染毒箱内,暴露于10支香烟中2次(对照组大鼠也置于染毒箱内做伪暴露),每次1 h,2次之间相隔不少于 4 h;开始燃烧4支香烟,随后每次2支香烟,每支香烟燃烧约12 min,换烟间歇3 min。吸烟组分别吸烟2个月和4个月;戒烟1个月组即大鼠吸烟4个月后暴露于常氧条件下1个月。
2实验试剂及仪器
芙蓉牌香烟(湖南中烟工业有限责任公司,尼古丁1.0 mg/支,焦油12 mg/支);TUNEL试剂盒购自武汉博士德生物公司;抗蛋白激酶R样内质网激酶(protein kinase R-like endoplasmic reticulum kinase, PERK)、p-PERK、真核起始因子(eukaryotic initiation factor, eIF)2α、p-eIF2α和CHOP抗体购自Santa Cruz;抗β-actin抗体、总蛋白提取试剂盒、BCA蛋白浓度检测试剂盒、SABC免疫组化试剂盒、辣根过氧化物酶标记的 II 抗、DAB显色剂和原位杂交试剂盒购自武汉博士德生物公司;地高辛标记的CHOP多相寡核苷酸探针购自北京鼎国生物有限公司;Western blot化学发光剂购自上海碧云天生物技术公司;逆转录试剂盒购自 MBI Fermentas;DNA Marker购自北京索莱宝生物科技有限公司。小动物肺功能测定系统和HX200型呼吸流量换能器购自北京新行兴业科贸有限公司;蛋白电泳系统购自Bio-Rad;PCR仪购自Eppendorf;PAS 900病理图像分析系统购自无锡朗伽生物工程有限公司;GIS-2010型凝胶成像分析系统购自上海天能科技有限公司;ELx800荧光酶标仪购自BioTek。CHOP的上游引物为5’-GCATCTTCATACACCACCACA-3’,下游引物为5’-TCATTCTCCTGCTCCTTCTC-3’;β-actin的上游引物为5’-GAGACCTTCAACACCCCAGC-3’,下游引物为5’-ATGTCACGCACGATTTCCC-3’。
3主要方法
3.1肺功能的测定 用小动物肺功能测定系统测定0.3秒用力呼气容积(forced expiratory volume in 0.3 second, FEV0.3)占用力肺活量(forced vital capacity, FVC)的比值(FEV0.3/FVC)和呼气峰流速(peak expiratory flow, PEF)。以1%戊巴比妥钠(40 mg/kg)腹腔注射麻醉大鼠,暴露气管,行气管插管,并与数据分析系统相连,在描记一段平静的呼吸后,在呼气末用注射器经三通管迅速打进6 mL空气(相当于深吸气),然后立即脱开,连接负压(-25 cmH2O, 1 cmH2O=0.098 kPa)抽气(相当于深呼气),经微机处理计算上述指标[3]。
3.2组织标本的留取 吸烟2个月组和4个月组大鼠末日吸香烟后,2 h内与对照组和戒烟组一起放血处死,将气管与双肺暴露,结扎右主支气管,用接12号针头(尖端磨平)的注射器经气管切开口向左肺注入4%多聚甲醛(含1‰ DEPC),至左肺复张后,结扎左主支气管。将右肺取出置于液氮中速冻,用于Western blot和RT-PCR;取出左肺浸入4%多聚甲醛(含1‰ DEPC)中固定,常规石蜡包埋。
3.3肺组织形态学的观察 取石蜡包块切片,厚度4 μm,每只大鼠选3张肺组织切片,二甲苯、乙醇脱蜡至水,苏木素浸染,盐酸乙醇分化,伊红溶液复染,再脱水透明封片,光镜下观察。
3.4TUNEL法检测肺组织中的凋亡细胞 取石蜡包块切片,厚度4 μm,每只大鼠选3张肺组织切片,二甲苯、乙醇脱蜡至水,加胃蛋白酶K于37 ℃消化20 min,滴加双氧水甲醇于室温30 min以灭活内源性酶,滴加50 μL的TUNEL 反应混合溶液,在湿盒中37 ℃ 孵育60 min,在孵育过程中覆盖盖玻片。按TUNEL检测试剂盒操作,DAB显色,阳性结果为棕黄色,PAS 900病理图像分析系统采集并分析图像。
3.5原位杂交检测肺组织中CHOP的mRNA表达 取石蜡包块切片,厚度4 μm,每只大鼠选3张肺组织切片,二甲苯、乙醇脱蜡至水,用3% H2O2封闭10 min,加20 μL预杂交液于湿盒内42 ℃恒温箱预杂交3.5 h;加20 μL杂交探针溶液,盖上原位杂交专用盖玻片置湿盒内,于40 ℃恒温箱内杂交20 h;在干燥环境中继续杂交2 h,洗涤、封闭,按原位杂交试剂盒说明书依次加入生物素化鼠抗地高辛、链酶亲和素-生物素-过氧化物酶复合物ABC,DAB显色,阳性结果为棕黄色。
3.6RT-PCR检测CHOP的mRNA表达 取右肺组织0.2 g,按说明书用TRIzol试剂提取总RNA,逆转录合成cDNA,以其为模板行PCR扩增。PCR反应条件为: 94 ℃预变性3 min; 94 ℃变性30 s, 72 ℃延伸5 min。CHOP的退火温度为56 ℃,扩增产物为432 bp;β-actin的退火温度为56 ℃,扩增产物为260 bp。取扩增产物5 μL经1.5%的琼脂糖凝胶电泳,用凝胶数字成像系统扫描分析扩增产物条带,分别测定各扩增带吸光度(A),将目的基因的扩增条带与内参照β-actin扩增带的吸光度比值作为其mRNA表达水平的相对指标。
3.7免疫组化检测肺组织中CHOP的蛋白表达 取石蜡包块切片,厚度4 μm,每只大鼠选3张肺组织切片,二甲苯、乙醇脱蜡至水,用3% H2O2封闭10 min,微波炉热抗原修复3次,每次10 min,冷却至室温后滴加封闭液,按SABC检测试剂盒操作。I抗用PBS稀释(CHOP 1∶100),孵育I抗, 4 ℃过夜。滴加相应II抗和SABC试剂, DAB显色,阳性结果为棕黄色。PAS 900病理图像分析系统采集和分析图像。
3.8Western blot法检测肺组织中PERK、p-PERK、eIF2α、p-eIF2α和CHOP的蛋白水平 于液氮中取出大鼠右肺组织,按照总蛋白提取试剂盒说明书提取总蛋白,并依据BCA蛋白浓度检测试剂盒说明书测定蛋白浓度。严格按照Western blot实验步骤进行操作。抗PERK、p-PERK、eIF2α、p-eIF2α、CHOP和β-actin I抗均按1∶500封闭液稀释, II 抗1∶1 000稀释。增强化学发光法X胶片显影定影后扫描至计算机,图像分析系统对扩增产物条带吸光度半定量分析。
4统计学处理
使用SPSS 13.0软件进行统计分析。计量资料数据采用均数±标准差(mean±SD)表示。多个样本比较采用单因素方差分析,组间两两比较采用SNK-q检验,以P<0.05为差异有统计学意义。
结 果
1大鼠肺功能的变化
与对照组比较,吸烟2个月组大鼠FEV0.3/FVC与PEF明显下降,吸烟4个月组肺功能较吸烟2个月组进一步下降,而戒烟组大鼠肺功能有所好转(P<0.01),见表1。
表1各组大鼠肺功能的变化
Table 1. Pulmonary function changes in each group (Mean±SD.n=10)

GroupFEV03/FVC(%)PEF(mL/s)Control83.47±4.9840.11±3.66CS⁃275.48±2.57∗33.42±3.21∗CS⁃466.18±4.12∗△25.89±2.22∗△Ex⁃S69.35±2.45∗#28.41±2.30∗#
*P<0.05vscontrol group;△P<0.05vsCS-2 group;#P<0.05vsCS-4 group.
2肺组织的病理学变化
与对照组相比,吸烟2个月组大鼠肺泡结构紊乱,支气管肺内出现以淋巴细胞为主的炎症细胞浸润,支气管壁杯状细胞数目增多,腺体增生肥大,部分肺泡出现破裂;吸烟4个月组大鼠肺泡结构紊乱更明显,支气管肺内有大量炎症细胞浸润,腺体增生肥大明显,肺泡壁破裂,肺泡腔扩大,部分融合成肺大泡;戒烟组大鼠肺泡腔扩大,较吸烟4个月组无显著改变,见图1。
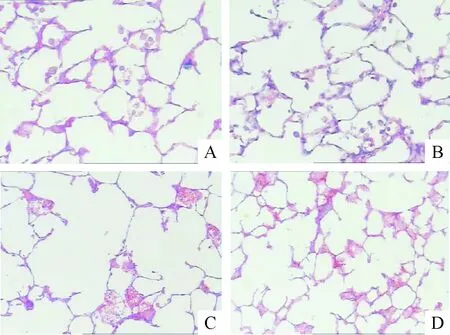
Figure 1. The morphological characters of the rat lung tissues in each group (HE staining, ×200). A: control group; B: CS-2 group; C: CS-4 group; D: Ex-S group.
图1各组大鼠肺组织的形态学改变
3COPD大鼠中的肺结构细胞凋亡
肺结构细胞的凋亡超过增殖是COPD的主要发病机制。TUNEL法结果发现,对照组、吸烟2个月组、吸烟4个月组和戒烟组大鼠肺结构细胞的凋亡率分别为(11.442±1.724)%、(22.400±1.615) %、(34.830±3.201)%和(31.360±1.820)%;凋亡的细胞主要是Ⅰ型肺泡上皮细胞、Ⅱ型肺泡上皮细胞、血管内皮细胞及支气管上皮细胞,见图2。
4肺组织中CHOP的mRNA表达
原位杂交结果光镜下见CHOP的mRNA主要表达于支气管和肺泡上皮细胞中,在对照组中弱表达,吸烟2个月组大鼠稍增强,在吸烟4个月大鼠表达最明显,呈黄色,在戒烟组大鼠和吸烟4个月大鼠表达无差异。RT-PCR检测结果示,吸烟2个月大鼠CHOP较对照组明显升高(P<0.05),吸烟4个月大鼠较2个月大鼠比较则进一步升高(P<0.05),而戒烟组大鼠与吸烟4个月大鼠相比较则无显著差异,与原位杂交的结果一致,见图3。
5大鼠肺组织中PERK、p-PERK、eIF2α、p-eIF2α及CHOP蛋白水平的变化
免疫组化光镜下见,CHOP主要在支气管、血管内皮细胞和肺泡上皮细胞的胞核中表达。其在对照组中弱表达,吸烟2个月组大鼠稍增强,在吸烟4个月大鼠表达最明显(P<0.05),呈黄色,在戒烟组大鼠和吸烟4个月大鼠表达无差异,见图4。Western blot结果显示,与对照组相比,吸烟2个月组大鼠p-PERK、p-eIF2α和CHOP的蛋白水平明显上升,在吸烟4个月组进一步显著升高(P<0.05),在戒烟组中稍下降,而PERK和eIF2α在各组蛋白水平的差异无统计学显著性,见图5。

Figure 2. Apoptotic cell death in the rat lung tissues after exposure to CS (×200). The sections of the rat lung tissues were prepared and subjected to TUNEL assay for visualizing apoptotic cells (yellow). A: control group; B: CS-2 group; C: CS-4 group; D: Ex-s group. Mean±SD.n=3.*P<0.05vscontrol group;△P<0.05vsCS-2 group.
图2各组大鼠的肺结构细胞凋亡
6肺功能、肺结构细胞凋亡率与CHOP蛋白及mRNA表达之间的相关性分析
肺功能(FEV0.3/FVC和PEF)与凋亡率之间呈负相关,与CHOP表达呈负相关(P<0.05); CHOP的mRNA与蛋白表达呈正相关,见表2。
讨 论
COPD是呼吸系统的常见疾病,其发病机制目前尚未完全阐明。近年来研究发现,除炎症、氧化应激和蛋白酶/抗蛋白酶失衡机制之外,肺结构细胞凋亡超过增殖导致肺结构不能维持是COPD发生发展的关键[4-6]。目前一般认为,香烟烟雾引起氧化应激是导致COPD肺泡上皮细胞凋亡的主要原因,但是关于氧化应激引起COPD肺泡上皮细胞凋亡的机制研究仍不多见。而内质网应激诱导肺上皮细胞的凋亡机制是目前COPD发病机制研究的重要内容[7]。本研究采用单纯被动吸烟法复制大鼠COPD模型,吸烟2个月大鼠肺功能较对照组大鼠明显下降,病理学检查结果显示肺结构紊乱,部分肺泡出现破裂,TUNEL结果显示吸烟2个月大鼠的凋亡细胞明显增加,而吸烟4个月后发现大鼠肺功能呈阻塞性通气障碍,表明大鼠吸烟4个月后COPD模型复制成功,肺泡结构紊乱,肺泡壁变薄,部分融合成肺大泡,大鼠组织病理学检查结果符合慢性支气管炎和阻塞性肺气肿改变,而TUNEL结果显示其凋亡率较吸烟2个月大鼠则进一步的增加。相关分析显示肺功能与凋亡率呈负相关,表明吸烟能导致肺结构细胞的凋亡,肺结构破坏不能维持其正常功能。而戒烟组的肺功能较吸烟4个月组稍下降,TUNEL结果显示其凋亡率和吸烟4个月组比较没有明显改变。吸烟是COPD发生发展的最重要危险因素,COPD的病理学表现为慢性支气管炎和阻塞性肺气肿,COPD的病理变化是不可逆的,戒烟是COPD的重要干预措施。推测戒烟后其气道炎症和肺气肿仍持续进展;戒烟只能减慢COPD进展的速度;而不能阻止COPD进展。
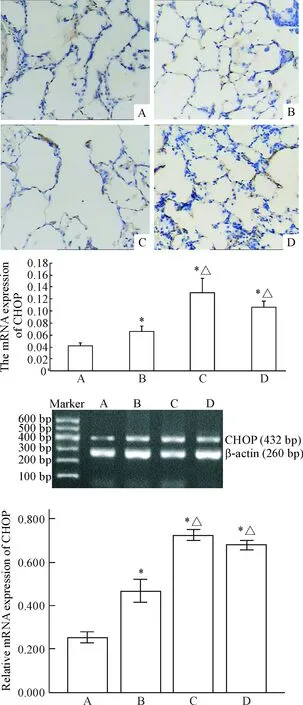
Figure 3. The distribution and expression of CHOP mRNA in rat lung tissues. The sections of the rat lung tissues were prepared and subjected toinsituhybridization for visualizing the distribution of CHOP mRNA in the cells and the mRNA was mainly located in nucleus (×200). The mRNA expression of CHOP was also detected by RT-PCR. A: control group; B: CS-2 group; C: CS-4 group; D: Ex-S group. Mean±SD.n=3.*P<0.05vscontrol group;△P<0.05vsCS-2 group.
图3肺组织中CHOPmRNA的分布和表达

Figure 4. The sections of the rat lung tissues were prepared and subjected to immunohistochemical staining for visualizing the distribution of CHOP protein (×200). A: control group; B: CS-2 group; C: CS-4 group; D: Ex-S group. Mean±SD.n=3.*P<0.05vscontrol group;△P<0.05vsCS-2 group.
图4肺组织中CHOP蛋白的表达
内质网(endoplasmic reticulum,ER)负责真核细胞内蛋白质(膜蛋白和分泌蛋白)的合成、加工、转运和钙离子浓度的调节。氧化应激、细胞毒性物质、缺氧、DNA损伤和病毒感染等许多因素可导致内质网腔内错误折叠、未折叠蛋白聚集和钙离子平衡紊乱,出现ERS,ERS状态下,内质网腔内错误折叠的蛋白质积聚增多,激活内质网膜上的跨膜感应蛋白,再通过其下游的信号调节通路介导细胞功能调节活动或启动细胞凋亡/自噬程序,称为未折叠蛋白反应(unfolded protein response,UPR)。哺乳动物UPR具有3条跨膜信号转导通路,分别由肌醇需求酶1(inositol-requiring enzyme 1,IRE1)、PERK和活化转录因子6(activating transcription factor 6,ATF6)执行跨膜信号转导[8-11]。活化的PERK迅速使eIF2α磷酸化,减少分泌蛋白的翻译以缓解内质网的负载。活化的IRE1具有核酸内切酶活性,剪接X盒结合蛋白1(X-box binding protein-1,XBP1)mRNA前体形成有活性的XBP1 mRNA,后者进一步翻译出具有功能活性的蛋白质。哺乳动物的ATF6包含α和β两种亚型,只有ATF6α参与UPR,水解活化的ATF6α进入细胞核与内质网应激反应元件结合并激活分子伴侣基因的表达,或者与XBP1结合形成异二聚体,再通过内质网应激反应元件激活内质网相关性蛋白降解相关基因和XBP1基因的表达。本研究的Western blot结果显示,PERK以及eIF2α在各组表达量的差异无统计学显著性,而p-PERK以及p-eIF2α则在对照组大鼠中的蛋白水平是最低的,在吸烟2个月大鼠中明显升高,在吸烟4个月大鼠中则进一步明显上升,而戒烟组大鼠则没有显著差别。由此得知吸烟可促进肺组织发生内质网应激。
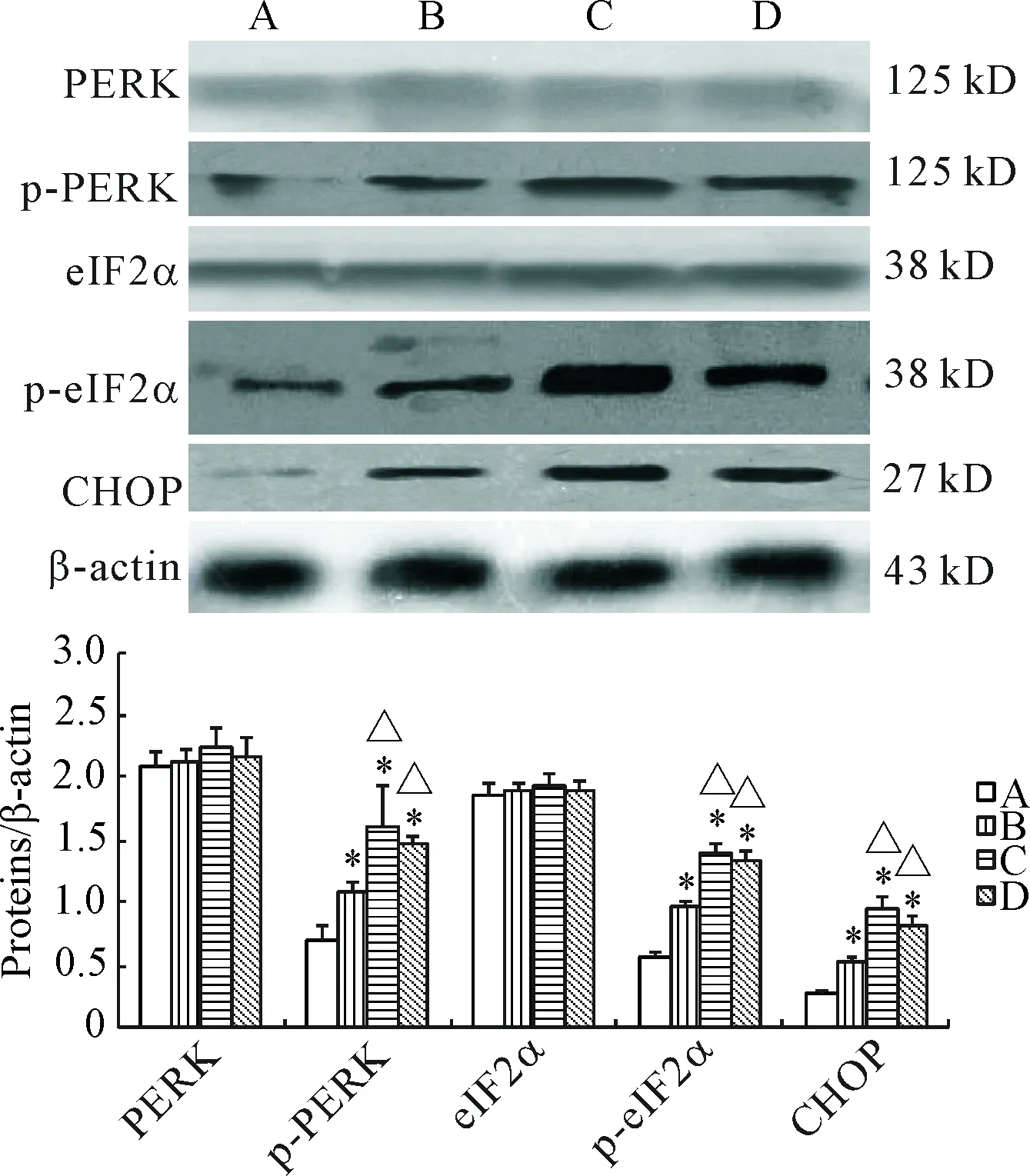
Figure 5. The relative protein expression of PERK, p-PERK, eIF2α, p-eIF2α and CHOP detected by Western blot. A: control group; B: CS-2 group; C: CS-4 group; D: Ex-S group. Mean±SD.n=3.*P<0.05vscontrol group;△P<0.05vsCS-2 group.
图5肺组织中PERK、p-PERK、eIF2α、p-eIF2α和CHOP的蛋白水平
表2肺功能、肺结构细胞凋亡率和CHOP表达之间的相关性分析
Table 2. Linear correlation analysis between different parameters of COPD rats
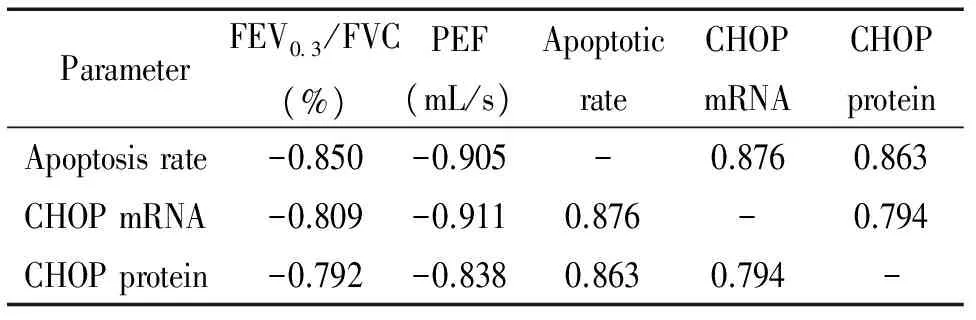
ParameterFEV03/FVC(%)PEF(mL/s)ApoptoticrateCHOPmRNACHOPproteinApoptosisrate-0.850-0.905-0.8760.863CHOPmRNA-0.809-0.9110.876-0.794CHOPprotein-0.792-0.8380.8630.794-
ERS是把双刃剑,应激时间短和轻中度应激的细胞选择适应性生存,而ERS时间过长或者应激过强引起细胞凋亡[4]。哺乳动物ERS诱导的凋亡主要有3条信号通路:CHOP信号通路、凋亡信号调节激酶1(apoptosis signal-regulating kinase 1,ASK1)-JNK信号通路和caspase-12信号通路[8, 10]。CHOP在正常生理状态下基本检测不到, 其是位于内质网腔的促凋亡分子,在内质网应激诱导凋亡中发挥重要作用,它阻断细胞增殖及通过调节其下游基因包括DOCs (downstream of CHOP)和caspase-1诱导内质网相关凋亡,也可抑制Bcl-2和上调Bax 水平介导细胞死亡,但在ERS状态下表达显著升高,是ERS的标志[8, 12]。内质网过度应激发生时,可通过PERK 介导的eIF2α磷酸化同时,也提升 mRNA与核糖体的结合效率,促进ATF4 的翻译,ATF4 则转入核后,诱导CHOP基因的表达,CHOP通过线粒体促进细胞凋亡。本实验通过吸烟2个月组大鼠p-PERK的蛋白水平增加,同时 CHOP蛋白表达上调,在吸烟4个月大鼠则更加明显。表明吸烟可引起肺结构细胞发生内质网应激,内质网应激诱导肺结构细胞凋亡,从而促进慢性阻塞性肺疾病的发生发展。
总之,本实验在动物层面研究证实了内质网应激诱导细胞凋亡参与了COPD的发生发展。然而香烟烟雾在细胞层面是否发生内质网应激以及是否有其它通路参与内质网应激诱导性凋亡需进一步研究。
[1] 甘桂香, 胡瑞成, 戴爱国. 内质网应激与慢性阻塞性肺疾病[J]. 国际呼吸杂志, 2010, 30(9):551-554.
[2] 王 慧, 程德云, 关 键. 单核细胞趋化蛋白-1与COPD大鼠气道炎症的研究[J]. 四川大学学报: 医学版, 2004, 35(5):634-637.
[3] 王梅芳, 戴爱国, 刘玉全. 血红素加氧酶1 调节γ谷氨酰半胱氨酸合成酶的表达在慢性阻塞性肺疾病大鼠肺组织中的作用[J]. 中国生物化学与分子生物学报, 2008, 24(12):1158-1164.
[4] Petrusca DN, Van Demark M, Gu Y, et al. Smoking exposure induces human lung endothelial cell adaptation to apoptotic stress[J]. Am J Respir Cell Mol Biol, 2014, 50(3):513-525.
[5] Lee KY, Park SY, Park S, et al. Progranulin protects lung epithelial cells from cigarette smoking-induced apoptosis[J]. Respirology, 2017, 22(6):1140-1148.
[6] Cantin AM, Richter MV. Cigarette smoke-induced proteostasis imbalance in obstructive lung diseases[J]. Curr Mol Med, 2012, 12(7):836-849.
[7] Yamada Y, Tomaru U, Ishizu A, et al. Decreased proteasomal function accelerates cigarette smoke-induced pulmonary emphysema in mice[J]. Lab Invest, 2015, 95(6):625-634.
[8] Chang CC, Kuan CP, Lin JY, et al. Tanshinone IIA faci-litates TRAIL sensitization by up-regulating DR5 through the ROS-JNK-CHOP signaling axis in human ovarian carcinoma cell lines[J]. Chem Res Toxicol, 2015, 28(8): 1574-1583.
[9] Roberson EC, Tully JE, Guala AS, et al. Influenza induces endoplasmic reticulum stress, caspase-12-dependent apoptosis, and c-Jun N-terminal kinase-mediated transforming growth factor-beta release in lung epithelial cells[J]. Am J Respir Cell Mol Biol, 2012, 46(5):573-581.
[10] 甘桂香, 胡瑞成, 戴爱国, 等. 吸烟慢性阻塞性肺疾病大鼠模型肺内内质网相关凋亡基因caspase-12表达研究[J]. 中国呼吸与危重监护杂志, 2011, 10(1):33-37.
[11] Fu P, Wu Q, Hu J, et al. Baclofen protects primary rat retinal ganglion cells from chemical hypoxia-induced apoptosis through the Akt and PERK pathways[J]. Front Cell Neurosci, 2016, 10:255.
[12] Jung KJ, Min KJ, Bae JH, et al.Carnosic acid sensitized TRAIL-mediated apoptosis through down-regulation of c-FLIP and Bcl-2 expression at the post translational levels and CHOP-dependent up-regulation of DR5, Bim, and PUMA expression in human carcinoma caki cells[J]. Oncotarget, 2015, 6(3):1556-1568.
