Mn基金属氧化物催化剂常温催化氧化甲醛
2018-01-23白梦天王广宏涛1赵云霞1陈敏东1南京信息工程大学江苏省大气环境与装备技术协同创新中心江苏南京10044南京信息工程大学环境科学与工程学院江苏南京10044南京信息工程大学江苏省大气环境监测与污染控制高技术研究实验室江苏南京10044
黄 琼,白梦天,任 超,王广宏,陶 涛1,,3,赵云霞1,,3,陈敏东1,,3 (1.南京信息工程大学江苏省大气环境与装备技术协同创新中心,江苏 南京 10044;.南京信息工程大学环境科学与工程学院,江苏 南京10044;3.南京信息工程大学江苏省大气环境监测与污染控制高技术研究实验室,江苏 南京 10044)
随着人民经济生活水平的提高,室内装饰已成为一种时尚,而被大量使用的装饰材料会缓慢释放出多种污染物,致使室内空气污染日趋严重,进而影响人体健康.甲醛是一种常见的室内空气污染物,具有毒性高,危害性大等特点.已经被世界卫生组织确定为致癌和致畸形物质[1-2].常用于甲醛净化的方法主要有吸附技术、光催化技术、低温等离子体技术以及常温催化氧化技术等[3],其中常温催化氧化技术是在较低的温度下将甲醛分解为无害的H2O和CO2,这种技术具有净化效率高、无二次污染,不存在吸附饱和等优点,且对低浓度甲醛污染亦有良好的净化效果,因此被认定为最有效的甲醛去除技术之一[4-5].
研究人员[6]采用共沉淀法研制了一系列贵金属Au/FeOx催化剂,当Au的负载量较低时,催化剂在常温条件下几乎无催化氧化甲醛活性,而当Au的负载量高达7.1wt%以上时,反应物转化率达到90%时的温度T90= 80℃.Alvarez-Galván[7]所研制的贵金属Pd/Al2O3催化剂在88℃条件下实现了甲醛的完全催化氧化.部分贵金属催化剂要达到 90%以上的甲醛净化效率均需要一定反应温度,而这难以满足室内常温低浓度甲醛的处理要求[8].因此开发常温条件下能将甲醛完全氧化为H2O和CO2的功能催化剂,具有重要的社会意义和经济价值.
目前常用于常温催化氧化甲醛的催化剂主要被分成两大类[9-10]:贵金属催化剂和非贵金属催化剂.贵金属催化剂研究主要集中在 Pt、Pd、Au、Ag 等贵金属,以其优异的氧化活性而常作为有机物催化氧化催化剂,具有活性高、稳定性好、耐高温等优点,值得注意的是Pt/TiO2已被研究证实是贵金属催化剂中较佳的常温催化氧化甲醛催化剂,中国科学院生态环境研究中心贺泓成功研制出能在常温条件下催化氧化甲醛的1wt% Pt/TiO2催化剂[11].研究发现,采用沉淀法制备的 MnOx-CeO2[12]催化剂在 100℃反应温度下实现了甲醛的完全催化氧化,而负载有贵金属的Pt/MnOx-CeO2[13]催化剂在常温条件下即实现完全催化氧化,表明贵金属具有极高的氧化活性,甚至常温条件下.由于贵金属催化剂成本高、资源稀缺,在催化氧化方面的应用受到很大限制.非贵金属催化剂主要有过渡、稀土金属氧化物和复合氧化物催化剂,在挥发性有机化合物(VOCs)净化处理方面具有重要的应用价值,且表现出较高的氧化性能及热稳定性[9].
本文采用络合法制备Mn-Ce-O室温催化氧化甲醛催化剂,并基于热处理术调控锰基金属氧化物晶体晶面,组装自发形成有序和异向生长的纳米晶体体系,应用正交实验设计方案,运用BET、XRD、SEM、TEM和IR等技术对催化剂性能及微观结构进行表征分析,以甲醛为探针反应物,考察Mn/Ce摩尔比、水化热温度、焙烧温度及焙烧时间等因素对催化剂催化氧化性能的影响,探索锰基金属氧化物价态、晶型、材料结构、形貌对催化剂常温氧化性能间的规律.
1 材料与方法
1.1 Mn-Ce-O室温催化氧化催化剂的制备
以硝酸铈和高锰酸钾为原料,通过络合法制备实验所需的 Mn-Ce-O混合氧化物催化剂.实验取 0.015mol硝酸铈溶于 40mL蒸馏水,再取0.015mol高锰酸钾溶于上述溶液中,在磁力搅拌器下搅拌溶解.将上述溶液转移到100mL聚四氟乙烯内胆不锈钢反应器中,并置于 120℃烘箱中反应 24h.待反应结束后取出反应物,经滤纸过滤、2次洗涤、4000r/min离心制得样品,并将样品置于90℃烘箱中烘干,经马弗炉 500oC, 4h焙烧处理,制得Mn/Ce摩尔比为1:1的催化剂,其它Mn/Ce 摩尔比(n(Mn)/n(Ce)=4:1, 1:2 和 1:4)、水化热温度(90,120,180℃)、焙烧温度(200,350,500℃)及焙烧时间(4,6,8h)催化剂均采用上述方法制备而成.
1.2 催化剂性能评价
催化剂性能评价在 0.2m3密闭玻璃反应装置中进行,通过甲醛降解效率来测定催化剂的活性.将甲醛溶液滴加至培养皿上,置入玻璃反应器内,待其充分挥发,使反应器内气态甲醛起始浓度保持在 1.0~1.1mg/m3之间,再将 1g粉末态催化剂均匀摊放至另一培养皿(Φ80mm)上,置入玻璃反应器内,将反应器密封后每隔12h用甲醛分析仪(PPM-400ST)采样分析反应器中残留的甲醛浓度.
1.3 催化剂的表征
为了考察催化剂微观结构在催化反应过程的作用,用AXSD8衍射仪对催化剂进行了XRD检测,选用Cu靶射线管,扫描速率为4°/min,2θ衍射角范围为10~80°;催化剂的比表面积、孔径大小及其分布情况采用以 N2吸附的 AutosorbiQ-AG-MP表面积分析仪检测;表面形貌采用日本Hitachi公司生产的SU1510扫描电镜检测分析.材料纳米尺度的结构、晶格面以及晶格间距由日本 JEOL 公司生产的 200kV场发射透射电子显微镜(JEM-2100F)监测分析.样品红外光谱特征峰由美国热电公司生产的傅里叶红外光谱仪(Nicolet iS5)检测分析.
1.4 数据处理
Mn-Ce-O混合氧化物催化剂常温催化氧化甲醛转化率按如下公式计算:
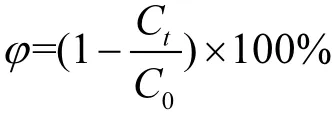
式中:Ct表示每12h后反应器内气态甲醛浓度;C0表示反应器内起始甲醛浓度.反应器内气态甲醛浓度每12h测试3次,取平均值.气态甲醛质量浓度直接由甲醛分析仪(PPM-400ST)直接读出.
2 结果与讨论
2.1 Mn-Ce-O混合氧化物常温催化氧化甲醛
图1(A)为未经焙烧处理Mn-Ce-O混合氧化物常温催化氧化甲醛活性,随着 Mn/Ce摩尔比的增加,其催化氧化活性持续下降,即 Mn/Ce摩尔比为4:1时,催化剂显示出最佳的氧化活性,其48h甲醛降解率为90.8%,而Mn/Ce摩尔比为1:4时,催化氧化活性最差,催化剂48h甲醛降解率仅为79.6%,且研究发现单组分MnOx所展示出的氧化活性差于Mn/Ce摩尔比大于1:1,而优于Mn/Ce摩尔比小于1:1.与此同时,实验对催化剂样品进行 500℃高温焙烧处理,其结果如图1(B)所示,双组分 Mn-Ce-O混合氧化物催化剂均展示出较未焙烧处理催化剂同等摩尔比条件下更佳的氧化活性,其中 48h Mn/Ce摩尔比为 4:1和 1:4时,甲醛转化率分别为 91.2%和92.1%,与中国科学院大连化学物理研究所申文杰采用沉淀法和浸渍法制备的 MnOx-CeO2[12]和 Pt/MnOx-CeO2[13]相比较,实现了非贵金属催化剂在常温条件下完全催化氧化.而对于单组分 MnOx催化剂,其氧化活性较未焙烧处理的MnOx催化剂活性显著降低,其48h甲醛降解率由 90.0%降低至 67.2%.单组分 CeO2仅表现出吸附性能,几乎无常温催化氧化活性,甲醛浓度24h后稳定于0.750mg/m3,效果较差.

图1 Mn-Ce-O混合氧化物催化剂常温催化氧化甲醛活性Fig.1 Catalytic oxidation activity of formaldehyde over Mn-Ce-O mixed oxide catalysts at room temperature
2.2 催化剂稳定性
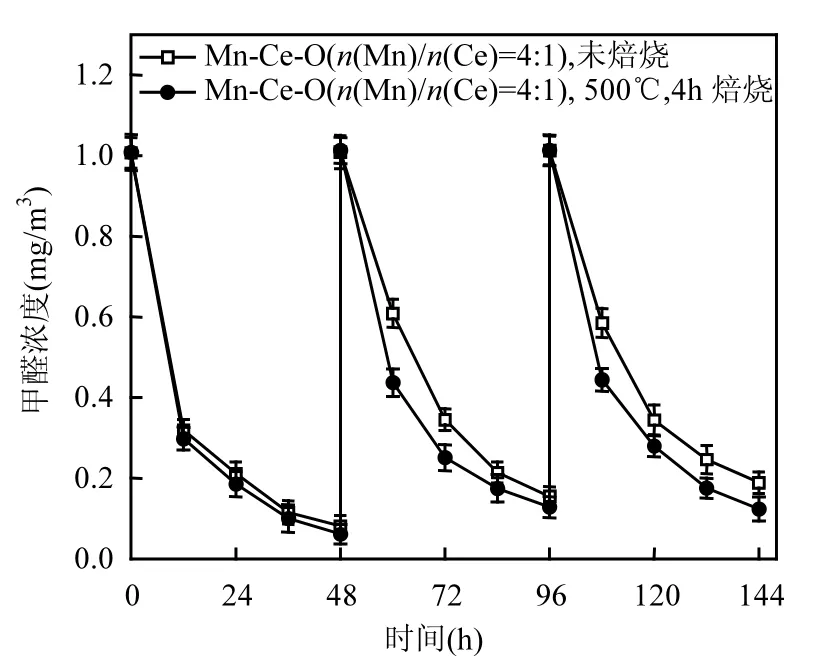
图2 Mn-Ce-O混合氧化物催化剂常温催化氧化甲醛稳定性Fig.2 Catalytic oxidation stability of formaldehyde over Mn-Ce-O mixed oxide catalysts at room temperature
实验对经焙烧处理(500℃,4h)和未经焙烧处理的同种 Mn-Ce-O混合氧化物催化剂常温催化氧化活性及稳定性进行了比较,如图2所示.由图2可知,未经焙烧处理的催化剂的氧化活性及稳定性均低于经焙烧处理的催化剂,且随着测试次数增加,其氧化活性逐渐降低,而经焙烧处理的 Mn-Ce-O混合氧化物催化剂经过3次常温催化氧化甲醛转化率分别为 93.9%,87.4%和 87.8%,其氧化活性虽略有下降,但整体氧化活性及稳定性明显优于未经焙烧处理的催化剂,因此本文后续实验均采用焙烧处理,以提高催化剂稳定性及氧化活性.
2.3 正交实验
基于前期实验基础,研究采用3因素3水平正交实验设计方案进一步优化催化剂制备条件[14],如表 1所示,设计了 A1B1C1; A1B2C2;A1B3C3; A2B1C2; A2B2C3; A2B3C1; A3B1C3;A3B2C1和A3B3C2 9组实验方案,以获得最佳的制备工艺参数,提高催化剂常温催化氧化性能.正交实验结果如表 2所示,表 2中的 K1、K2和 K3分别表示为各因素各水平下1g催化剂经36h反应后催化转化率总和,k1、k2和 k3分别表示为各因素各水平下催化剂经 36h反应后催化转化率平均值,在同一因素各水平下平均值的极差R(极差 R=最大平均值-最小平均值)反映各因素的水平变动对实验结果的影响大小,极差越大表示该因素的水平变动对实验结果越大,反之越小.

表1 三因素三水平正交实验设计Table 1 Three factors and levels selected for orthogonal experiment
研究发现,在同等实验条件下,上述9组催化剂均显示出较为良好的常温催化氧化活性,其中9#样品 36h甲醛降解率高达 94.2%.实验所得的极差由大到小的顺序依次为:RB>RA>RC,表明水化热温度是影响催化剂催化活性的最主要因素[15],主要因素应选择最佳水平,因而得到各因素的最佳配方为 A3B3C2,即为正交实验中 9#样品;n(Mn)/n(Ce)=1:1,水化热温度为 180℃,焙烧温度350℃.

表2 正交实验结果Table 2 Property results for orthogonal experimental
2.4 正交实验催化剂表征分析

表3 催化剂比表面积及孔容孔径Table 3 Physical properties of these catalysts in the orthogonal experiment
对正交实验中 9组样品进行比表面积及孔容孔径分析,如表3所示.测试发现,9组样品的平均孔径约为19.1nm,孔容约为0.065~0.085cm3/g,比表面积随着 Mn/Ce摩尔比由 1:4逐渐升高至1:1而逐渐降低,且 Mn/Ce摩尔比相同时,催化剂比表面积随着焙烧温度的升高而不断降低,由此可见,催化剂比表面积与Mn/Ce摩尔比和焙烧温度均成反比,其中Mn/Ce摩尔比为主要影响因素.当Mn/Ce摩尔比为1:4,焙烧温度为200℃时,催化剂显示最高的比表面积,达到 112.1m2/g,但该催化剂催化氧化活性最差,由此表明,比表面积不是催化剂氧化活性高低的主要因素,但与催化剂的晶型以及微观结构形态存在密切关系[16].

图3 正交实验样品XRD图Fig.3 XRD patterns of these catalysts in the orthogonal experiment
对正交实验中9组Mn-Ce-O混合氧化物催化剂晶型结构进行 XRD分析,如图 3所示.随着样品焙烧温度由 200℃升高至 500℃,尤其当Mn/Ce摩尔比为1:4时,样品中大量存在CeO2晶体(PDF 34-0394)衍射峰逐渐由宽峰演变成尖峰,2θ 分别为 28.5, 33.1, 47.5, 56.3°,表明 CeO2晶体结晶度随焙烧温度升高而增大,粒径增大.研究发现,仅当 Mn/Ce摩尔比为 1:1,且水化热温度设定为180℃时,XRD图谱分析中才发现有隐钾锰矿晶体(K-OMS-2,PDF 44-1386)衍射峰,其 2θ 分别为 28.7,37.4,41.6°[17].研究指出,Ce离子取代隐钾锰矿孔道中K+能使还原温度向低温方向移动,降低氧化温度,且 Ce离子取代改变了隐钾锰矿的吸收光谱,并且在全太阳光谱 200~ 2600cm-1波段范围内都具有强吸收,进而将光能转化为热能而有助于提高CeO2和隐钾锰矿混合催化剂常温氧化活性[18].
图4为正交实验中9组样品的扫描电镜图.当样品水化热温度为 90℃时,随着 Mn/Ce摩尔比由1:4升高至1:1,其样品表面呈现有越发增多的大量细小球状细颗粒物,且表面结构越蓬松.随着水化热温度的升高, 样品中块状物增多,其表面结构光滑,块状物结构越紧密.研究发现,随着 Mn/Ce摩尔比的升高,细小颗粒物越来越多, 其主要是因为水化热温度和氧化锰含量的增加,而与焙烧温度无关,实验表明影响催化剂结构的主要因素不是焙烧温度,而是水化热温度和 Mn/Ce摩尔比,这与正交实验结果描述相一致.


图4 正交实验样品SEMFig.4 SEM images of these catalysts in the orthogonal experiment

图5 实验样品HRTEM图Fig.5 HRTEM images of these catalysts in the orthogonal experiment
图5 为正交实验7#和9#样品的透射电镜图.由图5可知,当Mn/Ce摩尔比均为1:1时,经不同水化热温度处理的催化剂,其结构形貌迥然不同.当水化热温度设定为 90℃时,如图 5(A,B)所示,样品结构形貌主要以球状CeO2颗粒物形式存在,球状纳米粒子的平均粒径约 5nm,晶格面为CeO2的(1 1 1)面,晶格间距为0.30nm.与此同时,样品存有极少量的隐钾锰矿(K-OMS-2),晶格面为 K2-xMn8O16的(1 0 1)面,晶格间距为 0.65nm.9#样品中球状CeO2颗粒物较为分散的分布于大量纳米棒隐钾锰矿(PDF 44-1386)上,如图5(C,D)所示,其纳米棒直径约为 2~16nm,晶格面为隐钾锰矿的(2 0 0)面[19],晶格间距为0.50nm,与XRD分析结果相一致,Ce离子取代以及隐钾锰矿的还原能力和表面晶格氧活性在常温催化过程中起决定性的作用[20].同时在样品中发现有未分解的KMnO4,晶格面为 KMnO4的(2 0 0)面,晶格间距为0.456nm,这可能与过多的KMnO4未能完全溶解有关.

图6 实验样品红外光谱图Fig.6 Infrared spectrogram of these catalysts in the orthogonal experiment
图6 显示了正交实验中7#和9#样品常温催化氧化甲醛反应过程中表面物种的红外分析结果.在2925.5cm-1和2855.7cm-1出现的特征峰被归属为甲酸盐的两种不同取向的 C-H 的对称伸缩振动,在1384.9cm-1出现的特征峰被归属为甲酸盐的对称伸缩振动 νs(COO-)[21],3404.2cm-1处的宽峰和1612.7cm-1处的尖峰被归属为催化剂表面H2O,羟基自由基或含水物种的对称伸缩振动 νs(-OH),在 2426.9cm-1出现的特征峰被归属为 CO2的不对称伸缩振动[22].研究结果表明,作为反应的中间物种甲酸盐和最终产物CO2均被检测到, 其表明 Mn-Ce-O催化剂催化氧化甲醛确实可在常温下进行,且两种样品中均发现有 C-H键,这可能是已被吸附但尚未反应的甲醛.研究发现,7#样品的红外光谱特征峰明显高于9#样品,其吸水性更优,但氧化活性劣于9#样品,这可能是由于样品吸水性过强而导致的竞争吸附,催化氧化甲醛转化率降低,这与实验结果相一致.
2.5 焙烧时间对最佳配方催化剂性能的影响
研究对样品在 350℃下,经不同焙烧时间处理的晶型进行了XRD分析,如图7所示.随着焙烧时间的延长,CeO2晶体(PDF 34-0394)衍射峰逐渐增强,表明长时间焙烧处理可有效促使CeO2晶体结晶度增大,粒径增大.同时随着焙烧时间的延长,隐钾锰矿晶体(K-OMS-2)衍射峰亦逐渐增强,晶格缺陷逐渐减少.研究指出,增加K-OMS-2纳米棒氧空位缺陷浓度能够弱化Mn-O键,能够使K-OMS-2晶格氧的CO还原峰向低温方向移动,从而提高其晶格氧活性,因此延长焙烧时间因不利于 K-OMS-2纳米棒氧空位缺陷浓度增加而导致催化剂常温氧化活性逐渐降低[23].

图7 经不同焙烧时间处理的样品XRD图Fig.7 XRD patterns of these samples treated with different calcination time

对经不同焙烧时间处理的样品表面结构形态进行了SEM分析,如图8(A,B和C)所示,催化剂表面结构形态未发生显著变化,仍以块状物结构形式存在,无明显烧结熔融现象发生.
9#样品在 350℃下经不同焙烧时间处理后常温催化氧化甲醛的活性如图 9(A)所示.研究发现,随着焙烧的时间的延长,催化剂氧化活性逐渐降低,反应速率逐渐降低,且基于催化剂制备成本考虑,催化剂经350oC,4h的焙烧即可获得良好的催化氧化性能,36h甲醛降解率仍为 94.2%,而延长焙烧时间不利于催化剂氧化性能的发挥.实验并将最佳样品、活性炭吸附性能以及反应器稳定性进行了比较.由图9(A)可知,玻璃反应器密封性良好,36h甲醛损失率仅为 11.8%,其甲醛浓度最终稳定在 0.850mg/m3.在等量的活性炭吸附作用下,36h甲醛吸附率为 42.0%,表明活性炭确实存有吸附甲醛性能,但效果远不及 Mn-Ce-O混合氧化物催化剂.
2.6 最佳活性配方催化剂稳定性研究
通过对9#样品进行稳定性和重复性研究(如图 9(B)所示),发现随着催化降解次数的增加,甲醛降解速率略有下降,新鲜样品 36h即可将甲醛降至国标(0.08mg/m3)以下,而再经 3次重复性测试后,催化剂需要在48h将甲醛降至国标以下,且降解效率基本稳定,表现出良好的催化稳定性.
3 结论
3.1 影响催化剂催化活性的最主要因素是水化热温度,其次为Mn/Ce摩尔比,两者改变了催化剂的微观结构形态,而催化剂催化活性与比表面积和焙烧温度成反比,影响催化剂氧化活性高低的关键因素并非比表面积,而是催化剂的结构形态以及Mn-Ce-O混合氧化物中氧化物的相互作用.随着水化热温度的升高, 催化剂中的块状物增多,结构越紧密;随着Mn/Ce摩尔比的增加, 催化剂中细小球状颗粒物增多,表面结构越蓬松,其结构形态与催化剂正交实验结果相一致.
3.2 正交实验中 9#样品,即当 n(Mn)/n(Ce)=1:1,水化热温度为180℃,焙烧温度为350℃,4h时,催化剂表现出最佳的常温氧化活性,36h甲醛降解率为 94.2%,且稳定性良好,氧化活性并未随焙烧时间的延长而下降.
[1]Fan Z Y, Zhang Z X, Fang W J, et al. Low- temperature catalytic oxidation of formaldehyde over Co3O4catalystsprepared using various precipitants [J]. Chinese Journal of Catalysis, 2016,37(6):947—954.
[2]代 伟,周亚平,周 理.SBA-15介孔分子筛负载金属酞菁催化氧化甲醛 [J]. 中国环境科学, 2006,26(4):400—403.
[3]叶 青,闫立娜,罗才武,等. Au负载OMS-2催化剂的制备及其低温催化氧化性能 [J]. 中国环境科学, 2012,32(4):609—616.
[4]Wang L F, Sakurai M, Kameyama H. Study of catalytic decomposition of formaldehyde on Pt/TiO2alumite catalyst at ambient temperature [J]. Journal of Hazardous Materials, 2009,167(1):399—405.
[5]Kim S S, Park K H, Hong S C. A study on HCHO oxidation characteristics at room temperature using a Pt/TiO2Catalyst [J].Applied Catalysis A: General, 2011,398(1):96—103.
[6]Li C Y, Shen Y N, Jia M L, et al. Catalytic combustion of formaldehyde on gold/iron-oxide catalysts [J]. Catalysis Communications, 2008,9(1):355—361.
[7]Álvarez-Galván M C, Pawelec B, Peña O'Shea V A, et al.Formaldehyde/Methanol Combustion on Alumina-Supported Manganese-Palladium Oxide Catalyst [J]. Applied Catalysis B:Environmental, 2004,51(2):83—91.
[8]Huang H B, Leung Y C. Complete elimination of indoor formaldehyde over supported Pt catalysts with extremely low Pt content at ambient temperature [J]. Journal of Catalysis, 2011,280(1):60—67.
[9]Bai B Y, Qiao Q, Li J H, et al. Progress in research on catalysts for catalytic oxidation of formaldehyde [J]. Chinese Journal of Catalysis, 2016,37(1):102—122.
[10]An N H, Yu Q S, Liu G, et al. Complete oxidation of formaldehyde at ambient temperature over supported Pt/Fe2O3catalysts prepared by colloid-deposition method [J]. Journal of Hazardous Materials, 2011,186(1):1392—1397.
[11]Zhang C B, Li Y B, Wang Y F, et al. Sodium-Promoted Pd/TiO2for Catalytic Oxidation of Formaldehyde at Ambient Temperature[J]. Environment Science Technology, 2014,48(10):5816—5822.
[12]Tang X F, Li Y G, Huang X M, et al. MnOx—CeO2mixed oxide catalysts for complete oxidation of formaldehyde: Effect of preparation method and calcinations temperature [J]. Applied Catalysis B: Environmental, 2006,62(1):265—273.
[13]Tang X F, Chen J L, Huang X M, et al. Pt/MnOx—CeO2catalysts for the complete oxidation of formaldehyde at ambient temperature [J]. Applied Catalysis B: Environmental, 2008,81(1):115-121.
[14]黄 琼,马文娇,严小康,等.堇青石负载稀土铈基催化剂催化燃烧苯的性能研究 [J]. 功能材料, 2012,43(22):3079—3083.
[15]Zhou L, Zhang J, He J H, et al. Control over the morphology and structure of manganese oxide by tuning reaction conditions and catalytic performance for formaldehyde oxidation [J]. Materials Research Bulletin, 2011,46(10):1714—1722.
[16]Ma L, Wang D S, Li J H, et al. Ag/CeO2nanospheres: Efficient catalysts for formaldehyde oxidation [J]. Applied Catalysis B:Environmental, 2014,148—149(6):36—43.
[17]Tian H, He J H, Liu L L, et al. Effects of textural parameters and noble metal loading on the catalytic activity of cryptomelane-type manganese oxides for formaldehyde oxidation [J]. Ceramics International, 2013,39(1):315—321.
[18]侯静涛,李远志. MnO2基催化剂的微结构调控及其催化净化VOCs性能 [D]. 武汉:武汉理工大学, 2014.
[19]杨振前,胡建强.银/二氧化锰纳米材料的制备及其甲醛催化氧化性能研究 [D]. 广州:华南理工大学, 2015.
[20]Li H J, Qi G S, Tana, et al. Low-temperature oxidation of ethanol over a Mn0.6Ce0.4O2mixed oxide [J]. Applied Catalysis B:Environmental, 2011,103(1):54—61.
[21]庞光龙,张韫宏,麻春艳. MnOx基催化剂上甲醛室温催化氧化反应的研究 [D]. 北京:北京理工大学, 2015.
[22]Huang Q, Ma W J, Yan X K, et al. Photocatalytic decomposition of gaseous HCHO by ZrxTi1-xO2catalysts under UV—vis light irradiation with an energy-saving lamp [J]. Journal of Molecular Catalysis A: Chemical, 2013,366(1):261—265.
[23]Hou J T, Li Y Z, Liu L L, et al. Effect of giant oxygen vacancy defects on the catalytic oxidation of OMS-2 nanorods [J]. Journal of Materials Chemistry A, 2013,1(23):6736—6741.
