马西替坦片中有关物质的HPLC测定
2018-01-05聂忠莉胡一冰王晓玲萧茂玲
聂忠莉,王 倩,胡一冰,王晓玲,叶 丁,张 勇,萧茂玲,郭 瑞
(1.成都大学 药学与生物工程学院,四川 成都 610106;2.成都克莱蒙医药科技有限公司,四川 成都 610041)
马西替坦片中有关物质的HPLC测定
聂忠莉1,王 倩1,胡一冰1,王晓玲2,叶 丁2,张 勇2,萧茂玲2,郭 瑞2
(1.成都大学 药学与生物工程学院,四川 成都 610106;2.成都克莱蒙医药科技有限公司,四川 成都 610041)
为测定马西替坦片中的有关物质,利用高效液相色谱法,用十八烷基硅烷键合硅胶为填充剂,以水(1 000 mL水,加入三乙胺1 mL,用磷酸调至pH值=3.0)为流动相A,乙腈为流动相B,梯度洗脱,检测波长为220 nm,柱温40 ℃,流速1.0 mL/min,进样量10 μL.实验显示,在各破坏条件下产生的杂质与主峰以及杂质与杂质之间分离度均在1.5以上,能达到完全分离,杂质MAA6-1、MAA6-2、MAA7-3、MAA6检测限分别为0.019 ng、0.013 ng、0.072 ng、0.108 ng,峰面积与浓度呈良好的线性关系,校正因子都为1.0.结果表明,本方法简单、准确度高,专属性强,适用于马西替坦片中有关物质的测定.
马西替坦;有关物质;高效液相色谱法
0 引 言
肺动脉高压,是一种以肺血管阻力升高最终导致右心衰竭的严重的症候群[1].马西替坦,为美国食品药品管理局(FDA)批准用于治疗肺动脉高压(PAH)的一类内皮素受体拮抗剂[2].内皮素受体拮抗剂类药物能使肺动脉松弛、降低肺部血压[3].临床实验证明,马西替坦的耐药性和安全性良好,药物与药物之间相互作用风险较小,可降低肺动脉高压(PAH)患者的发病率和死亡率,且主要是通过减缓疾病进展发挥作用[4-5].马西替坦片为马西替坦的制剂,而制剂质量标准中有关物质项下主要考虑的杂质为原料在储存和运输过程中产生的降解杂质,以及在制剂制备工艺过程与储存及运输过程中产生的降解杂质.本研究拟采用高效液相方法,对本品的杂质MAA6-1、MAA6-2、MAA7-3和MAA6进行分析,并对其进行了方法学验证,拟对制剂的质量控制提供相应的依据.
1 试药与仪器
1.1 试 药
实验所用试药包括:马西替坦对照品(批号,130802),杂质MAA6-1对照品(批号,MAA6-1-20130601),杂质MAA6-2对照品(批号,MAA6-2-20130601),杂质MAA7-3对照品(批号,MAA7-3-20130701),杂质MAA6对照品(批号,A6-130726),马西替坦片(小试3批的批号:140101、140102、140103),均由成都克莱蒙医药科技有限公司提供;马西替坦片(中试3批批号,140201、140202、140203),由峨眉山通惠制药有限公司生产,马西替坦片(批号,UM 010B0801),由Actelion Pharmaceuticals,Ltd生产;乙腈(色谱纯)、磷酸(分析纯)、三乙胺(分析纯),由天津科密欧公司生产.
1.2 仪 器
实验所用仪器包括:5410/L-2455型高效液相色谱仪(Hitachi HITACHI),BP 210D型电子天平(Sartorius),pHS-3C型酸度计(上海佑科).
2 方法与结果
2.1 色谱条件
实验的色谱条件为:用十八烷基硅烷键合硅胶为填充剂,以水(1 000 mL水,加入三乙胺1 mL,用磷酸调至pH值为3.0)为流动相A,乙腈为流动相B,梯度洗脱,检测波长为220 nm,柱温40 ℃,流速1.0 mL/min,进样量10 μL.梯度洗脱程序见表1.

表1 梯度洗脱程序
2.2 溶液配制
取本品适量,精密称定,加乙腈溶解并定量稀释至每1 mL约含1.0 mg的溶液,超声15 min,作为供试品溶液.
精密量取供试品溶液适量,加乙腈制成每1 mL中约含1 μg的溶液,作为对照溶液.
精密称取杂质MAA6-1、MAA6-2、MAA7-3、MAA6和马西替坦对照品适量,用乙腈溶解并稀释制成每1 mL含各杂质约1.0 μg、含马西替坦约1.0 mg的混合溶液,作为对照品溶液.
2.3 系统适用性
精密量取对照品溶液10 μL,注入液相色谱仪,记录色谱图,MAA6-2、MAA6-1、MAA6、MAA7-3和马西替坦分别依次出峰,各成分峰之间的分离度应符合规定,理论塔板数按马西替坦主峰计不得低于5 000.
精密量取对照溶液10 μL注入液相色谱仪,调节检测灵敏度,使主成分色谱峰峰高约为满量程的20%;再精密量取供试液、对照溶液和对照品溶液10 μL注入液相色谱仪,记录色谱图至主成分峰保留时间的2倍.
供试品溶液色谱图中如有与对照品溶液峰保留时间一致的峰,按外标法以峰面积计算,不得过0.1%;其他杂质峰的峰面积不得大于对照液主峰面积的0.1倍(0.1%),总杂质不得超过0.5%.
2.4 专属性
精密称取马西替坦样品适量分别进行强酸、强碱、氧化、高温和光照破坏性实验.取上述专属性试验样品溶液,分别进样,测定结果见图1和表2.
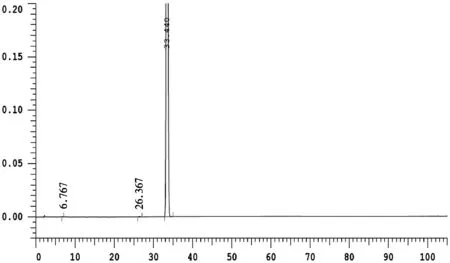
(a)未破坏样品图谱

(b)酸破坏样品图谱
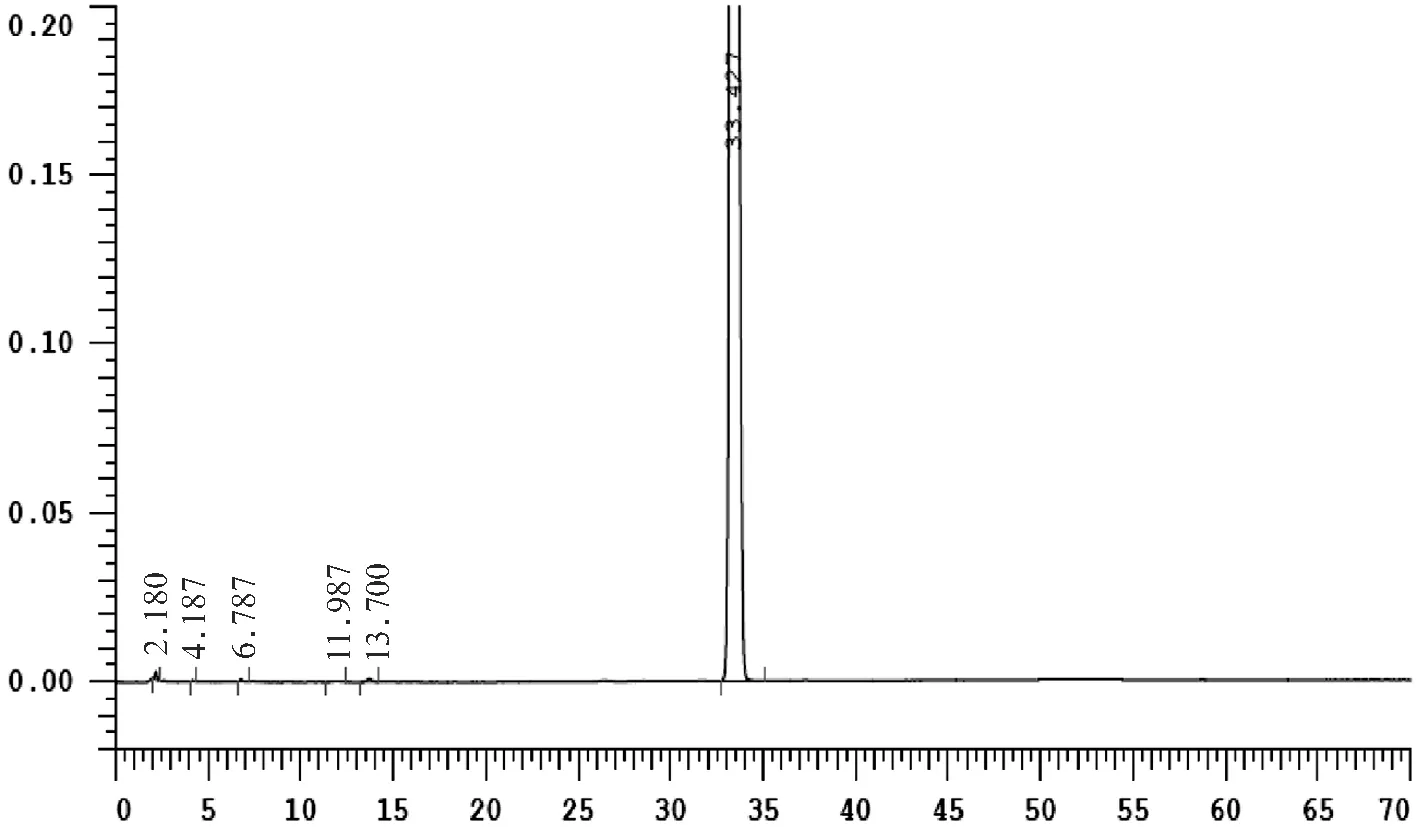
(c)碱破坏样品图谱

(d)氧化破坏样品图谱

(e)高温破坏样品图谱

(f)光照破坏试验图谱

(g)杂质对照品图谱
图1样品实验图谱
从图1可知,各峰依次为杂质MAA7-8、MAA6-2、MAA6-1、MAA7-6、起始物料C、中间体A6、MAA7-7、MAA7-2、MAA7-3、中间体A5、起始物料A4和马西替坦。
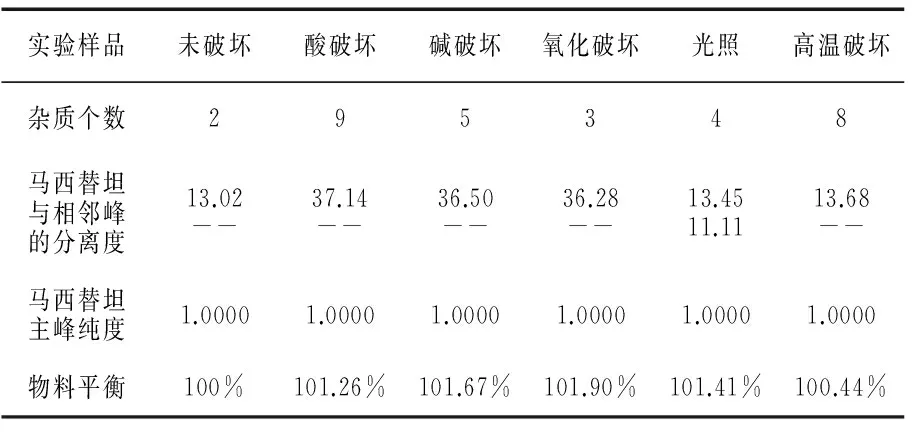
表2 有关物质专属性实验结果
由专属性实验结果可知,在各破坏条件下产生的杂质与主峰,以及杂质与杂质之间分离度均在1.5以上,能达到完全分离,主峰峰纯度均达到1.0000,杂质峰峰纯度均达0.99以上,峰的降解平衡在90%~102%之间.实验结果表明,方法专属性良好,能检测出本品在储藏和运输过程的极端条件下产生的降解产物.
2.5 重复性
取马西替坦自制片10片,精密称定,研细,精密称取细粉适量(相当于马西替坦10 mg)置10 mL量瓶中,加入杂质贮备液(含杂质约为0.1 mg/mL)各0.1 mL,加乙腈至接近刻度,超声提取15 min,取出,放冷至室温,用乙腈定容至刻度,摇匀,滤过,取续滤液作为供试品溶液,同法制备6份样品.
取上述供试品溶液10 μL,注入液相色谱仪,记录色谱图,按外标法以峰面积计算各杂质的含量,同一样品连续重复测定6次,各杂质含量的RSD%均小于2.0%,表明方法的重复性良好.
2.6 稳定性
取马西替坦自制片10片,精密称定,研细,精密称取细粉适量(相当于马西替坦10 mg),置10 mL量瓶中,加入杂质贮备液(含杂质约为0.1 mg/mL)各0.1 mL,加乙腈至接近刻度,超声提取15 min,取出,放冷至室温,用乙腈定容至刻度,摇匀,滤过,取续滤液作为供试品溶液.
取上述供试品溶液分别于0、2、4、8、24 h分别测定,记录图谱,以各成分峰面积计算RSD.实验结果表明,供试品溶液在室温下放置24 h,其峰面积和杂质个数均无明显变化,供试溶液室温放置24 h稳定.
2.7 检测限与定量限
取马西替坦及各已知杂质适量,用乙腈溶解后,分别稀释制成适宜浓度,分别精密吸取一定量注入液相色谱仪并检测信号噪音比.检测结果为:MAA6-1、MAA6-2、MAA7-3、MAA6检测限分别为0.019 ng、0.013 ng、0.108 ng、0.072 ng,定量限分别为0.116 ng、0.052 ng、0.251 ng、0.162 ng.定量限样重复6次,进样峰面积RSD分别为2.57%、4.21%、1.70%、3.64%.
2.8 线 性
精密称取马西替坦及各杂质适量,用乙腈逐级稀释,制备系列混合溶液(0.05 μg~1.5 μg/mL)并分别进样测定,记录色谱图,以进样浓度对峰面积进行线性回归,求得标准曲线.实验结果表明,杂质MAA6-1在浓度为0.05~1.5 μg/mL范围内,进样10 μL,峰面积与浓度呈良好的线性关系,其线性方程式为,Y=143 655.59x+1 099.39,R2=0.9997,校正因子1.0;杂质MAA6-2在浓度为0.05~1.6 μg/mL范围内,进样10 μL,峰面积与浓度呈良好的线性关系,其线性方程式为,Y=146 467.46x-1 067.70,R2=0.9987,校正因子1.0;杂质MAA7-3在浓度为0.06~1.9 μg/mL范围内,进样10 μL,峰面积与浓度呈良好的线性关系,其线性方程式为,Y=159 007.60x-587.41,R2=0.9998,校正因子1.0.杂质MAA6在浓度为0.05~1.5 μg/mL范围内,进样10 μL,峰面积与浓度呈良好的线性关系,线性方程式为,Y=138 620.14x+510.81,R2=0.9999,校正因子1.0.
2.9 回收率
1)杂质对照溶液的制备.分别取马西替坦各杂质适量,精密称定,分别用乙腈配制成每1 mL约含0.1 mg的贮备溶液.
2)对照品溶液的制备.分别取杂质贮备液各0.1 mL,置同一10 mL量瓶中,用乙腈稀释至刻度,摇匀,作为对照品溶液.
3)供试品溶液的制备.取马西替坦粉末适量(相当于马西替坦10 mg),共10份,精密称定,分别置10 mL量瓶中,其中1~3号样品分别加入杂质贮备液0.05 mL(相当于杂质标准限度50%),4~6号样分别加入杂质贮备液0.1 mL(相当于杂质标准限度100%),7~9号样品分别加入杂质贮备液0.15 mL(相当于杂质标准限度150%),再分别加入乙腈至近刻度,超声提取15 min,取出,放冷至室温,用乙腈定容至刻度,摇匀,滤过,取续滤液,作为供试品溶液.
按有关物质检查色谱条件分别精密吸取上述10 μL进样,注入液相色谱仪,记录色谱图,按外标法计算各样品中各杂质的含量和回收率.实验结果显示,杂质MAA6-1、MAA6-2、MAA7-3、MAA6的平均回收率分别为,102.68%、101.74%、105.47%、104.00%,RSD分别为,1.50%、1.18%、1.51%、1.07%.
实验结果表明,各杂质的回收率均在100%~110%之间,RSD均小于2.0%.表明本方法回收率良好,准确度符合要求.
2.10 精确度
由不同专业人员在不同日期用不同仪器按重复性试验测定6份供试液有关物质,记录色谱图,计算结果的RSD.实验结果表明,同一批样品在不同日期由不同专业人员在不同仪器上测得含量基本一致,RSD%小于2.0%.表明本方法精密度符合要求.
2.11 耐用性
取本品粉末适量,精密称定,置10ml量瓶中,加入杂质对照品贮备液0.1 mL,加入乙腈至近刻度,超声提取15 min,取出,放冷至室温,用乙腈定容至刻度,摇匀,滤过,取续滤液,制得供试品溶液.
在变动色谱条件中的流动相pH值、流速、柱温及不同色谱柱条件下,分别精密量取供试品溶液10 μL注入液相色谱仪,记录色谱图,考察各杂峰之间、杂峰与主峰之间的分离情况及各杂质含量的变化情况,结果见表3.

表3 有关物质耐用性实验结果
表3结果显示,在测定条件出现小的变动时,所测定的各杂质含量变化不大,含量的RSD%均小于5.0%.表明本方法的耐用性良好.
2.12 样品测定
按照上述色谱条件,采用高效液相色谱法对小试和中试各批样品及对照药品进行测定,测定结果见表4.

表4 小试、中试样品及对照药品检测结果
结果表明,在3批小试、3批中试样品中均只检验出了降解杂质MAA7-3.
3 结 论
本研究参照《中国药典》2015年版中药品质量分析方法验证指导原则,对该方法进行了方法学验证.结果表明,方法学验证项目中各项均符合检测要求.最终确定马西替坦片中有关物质检测条件为:采用高效液相色谱法,用十八烷基硅烷键合硅胶为填充剂,以水(1 000 mL水,加入三乙胺1 mL,用磷酸调至pH值为3.0)为流动相A,乙腈为流动相B,梯度洗脱,检测波长为220 nm.柱温40 ℃,流速1.0 mL/min,进样量10 μL.实验结果表明,本方法操作简便,高效快速,专属性好,灵敏度高,分离效果好,适用于马西替坦片中有关物质的检测.
[1]中国医学会心血管病学分会,《中华心血管病杂志》编辑委员会.肺动脉高压筛查诊断与治疗专家共识[J].中华心血管病杂志,2007,35(11):979-987.
[2]摘自《丁香园》.FDA批准马西替坦用于治疗肺动脉高血压[J].上海医药,2013,34(21):61-62.
[3]潘晓菲,谭初兵,时丽丽,等.新型肺动脉高压治疗药物马西替坦[J].中国新药杂志,2014,23(1):3-5.
[4]金玉洁,肖桂芝,王文倩,等.新型内皮素受体拮抗剂马西替坦[J].现代药物与临床,2013,28(3):405-407.
[5]赵文丽.马西替坦Ⅲ期临床试验资料表明治疗肺动脉高压有效[J].国际药学研究杂志,2016,40(6):806-80?.
[6]国家药典委员会.中华人民共和国药典[S].北京:中国医药科技出版社,2015.
DeterminationofRelatedSubstancesinMacitentanbyHPLC
NIEZhongli1,WANGQian1,HUYibing1,WANGXiaoling2,YEDing2,ZHANGYong2,XIAOMaoling2,GUORui2
(1.School of Pharmacy and Bioengineering, Chengdu University, Chengdu 610106, China; 2.Chengdu Climb Pharmaceutical Technology Co., Ltd., Chengdu 610041, China)
The paper establishes an HPLC method for the determination of the related substances in macitentan.The silane bonded silica is used as filler,with water(1 000 mL water,with three triethylamine 1ml being added,adjusted to pH 3.0 with phosphoric acid) as mobile phase A,acetonitrile as mobile phase B,Gradient elution.The detection wavelength is 220 nm.With the flow rate of the mobile phase being 1.0 mL/min and column temperature being 40 ℃,the sample volume is 10 μL.The results show that the separation degree between the main peak and impurity produced in different damaging conditions,as well as that between impurity and impurity is more than 1.5,which means that complete separation can be achieved.The detection limits of impurity MAA6-1,MAA6-2,MAA7-3 and MAA6 are 0.019,0.013,0.072,0.108 ng respectively.There is a good linear relationship between the peak area and the concentration.The correction factor is 1.0.It's concluded that the method mentioned in this paper is simple,accurate,specific enough and suitable for the determination of the related substances in macitentan.
macitentan;related substances;HPLC
TQ460.7+2;O657.7+2
A
1004-5422(2017)04-0342-05
2017-09-24.
聂忠莉(1966 — ),女,副主任药师,从事药物制剂与分析研究.
