耐铜植物茵陈蒿根际细菌群落结构及影响因素
2018-01-03邵宗圆杨如意
邵宗圆,王 悦,张 菊,杨 程,周 刚,杨如意
安徽师范大学环境科学与工程学院, 芜湖 241002
耐铜植物茵陈蒿根际细菌群落结构及影响因素
邵宗圆,王 悦,张 菊,杨 程,周 刚,杨如意*
安徽师范大学环境科学与工程学院, 芜湖 241002
采用MiSeq高通量测序技术对耐铜植物茵陈蒿根际的细菌16S rDNA基因V3—V4区片段进行了测序,研究了细菌群落结构的变化,并分析了其与土壤环境因子的关系。研究表明,采样点Cu3中细菌群落的多样性、丰富度、均匀度、ACE指数、Chao1指数等均显著低于Cu1和Cu2,但Cu3的覆盖度高于Cu1。排名前10的优势细菌门总相对丰度均在95%以上,其中8个优势细菌门在3个采样点中是相同的,包括Proteobacteria(变形菌门)、Acidobacteria(酸杆菌门)、Bacteroidetes(拟杆菌门)、Gemmatimonadetes(芽单胞菌门)、Actinobacteria(放线菌门)、Verrucomicrobia(疣微菌门)、Planctomycetes(浮霉菌门)和Unclassified(未分类门)等。采样点Cu1中变形菌门、拟杆菌门和芽单胞菌门的相对丰度显著高于其他两个采样点,而酸杆菌门、放线菌门、疣微菌门、绿弯菌门(Chloroflexi)和未分类门则刚好相反,表明细菌对胁迫环境的适应能力有明显差异。主坐标和冗余分析表明,3个采样点的细菌群落结构发生了明显改变。土壤环境因子与细菌群落的变化关系密切,8个因子的特征值共解释了97.5%的总方差。其中,总铜、总磷、pH、有效磷和有机质为显著性因子,可以解释93.9%的群落变化,但影响不同采样点细菌群落的主导因子有所差异。
重金属污染土壤;植物修复;根际细菌;群落结构;高通量测序
由于矿产的开采与冶炼、大气沉降、污灌、肥药使用和工业排放的影响,导致土壤重金属污染越来越严重。我国耕地土壤的点位超标率达19.4%,主要以镉、镍、铜等重金属无机污染为主[1]。目前,土壤重金属污染已成为全球性的环境问题之一,重金属污染土壤的修复、治理与复垦已经成为环境科学和生态学研究的热点领域。其中,植物修复作为一种原位修复技术,不仅处理成本低、环境友好,而且具有多重生态功能,受到国内外学者的高度关注[2- 3]。但植物修复也存在处理周期长、适用范围窄、处理效率低等缺陷,而且有可能通过取食作用增加植食性动物面临的生态风险[4- 5]。近年来,利用植物和微生物之间的相互作用,建立植物-微生物联合修复体系,强化植物修复效率成为重要的发展趋势[6- 7]。
微生物是联系植物与土壤环境的重要纽带,对两者间的相互作用具有重要的调节功能。微生物及其产生的各种代谢物广泛参与土壤、沉积物等环境介质中的物理、化学和生物化学反应,影响重金属的迁移、转化和生物有效性,进而影响植物的生长和对重金属的吸收与积累[6, 8- 10]。一方面,土壤微生物可以吸附或聚集重金属[11],从而降低重金属对植物的胁迫。 另一方面,微生物释放的有机物能够络合和浸出重金属,提高其生物有效性[12]。 研究发现,在氨基酸等有机物的作用下,纳米氧化铜浸出的Cu2+高达60%—75%,从而更易于被植物所吸收[13]。海州香薷(ElsholtziasplendensNakai)根际的耐铜细菌通过分泌有机酸,增加了铜的生物有效性,显著提高了植物对铜的累积水平,缩短了修复周期[14]。微生物不仅可以改变重金属形态,而且可以影响其粒径大小。研究表明,多种耐硒细菌可以将高浓度的硒酸盐或亚硒酸盐还原为纳米级单质硒,从而降低其生物毒性,可应用于硒污染土壤或水体的修复[15]。
微生物在重金属植物修复中的作用因具体的微生物、植物和重金属类型,以及环境条件的差异而有所差别。目前,大多数研究主要通过从环境介质中分离、筛选、驯化等方式对特定功能的少数微生物加以利用。但是,现实环境中微生物是以群落的形式存在的,微生物间的相互作用对植物修复效率也具有重要影响[16]。因此,研究植物根际的微生物群落,分析影响其群落结构的关键环境因子,对更好地发挥微生物的功能具有重要意义。
茵陈蒿(ArtemisiacapillariesThunb.)是一种传统的中药材,也是铜矿废弃地次生演替过程中的先锋植物,具有较强的铜耐性[2, 4],根部铜积累量高达533 mg/kg[17]。目前,国内外关于茵陈蒿根际微生物群落,以及在铜矿废弃地修复方面的相关研究还很少。本研究通过高通量测序法,研究了茵陈蒿根际的细菌群落结构,分析了细菌群落与土壤环境因子间的关系,本研究结果将为阐明细菌群落变化的关键驱动因子,更好地利用功能细菌强化铜矿废弃地的植物修复提供参考[16]。
1 材料与方法
1.1 采样点概况和样品采集
本研究采样点位于安徽省芜湖市南陵县大工山——凤凰山古铜矿区的一处铜矿废弃地(30°55′51″N, 118°9′23″E)。该地区属北亚热带湿润型季风气候区,东亚季风盛行,年均降雨量约1400 mm,年均蒸发量约1377 mm,年平均气温约15.8℃。该铜矿废弃地废弃时间超过50年,土壤为发育于坡积母质的棕红壤,质地属沙质土壤,极易发生水土流失。但是,表层土由于长期自然演替导致腐殖质含量较高。在研究点内,植被类型以草本植物为主(占40%以上),海州香薷、鸭跖草(CommelinacommunisL.)、茵陈蒿、酸模(RumexacetosaL.)和头花蓼(PolygonumcapitatumBuch.-Ham. ex D. Don)等为优势种,零星散布着少量枸骨冬青(IlexcornutaLindl.etPaxt)和白花泡桐(Paulowniafortunei(Seem.) Hemsl.)等灌木和乔木[4]。
本研究以3个面积约为20 m2的废弃地作为采样点,样点之间相距约100—120 m。每个采样点随机选取4个1 m × 1 m 的样方(每个样方作为一个重复)。将样方内的茵陈蒿整株取出,采用抖根法收集根际土壤,同一样方内的土样混合均匀,作为一个混合样(约200 g)。每个采样点的4个样品不混合,共计12个样品。所有土壤样品立即带回实验室,部分新鲜土壤用于提取细菌总DNA,其余土壤自然风干后待用。
1.2 试验方法
1.2.1土壤理化性质测定
土壤pH的测定:称取过1mm筛孔的风干土10g,加入1mol/L的KCl溶液25mL(土水比为1∶2.5),摇匀30min后用pH计进行测定。土壤总磷和有效磷测定:分别采用H2SO4-HClO4消解和HCl-H2SO4浸提,钼铵蓝比色法进行测定。有机质的测定采用K2Cr2O7-H2SO4稀释热法[18]。土壤Cu、Zn总量采用HF-HClO4-HNO3消解,有效态Cu、Zn通过0.1mol/L的HCl浸提[18],通过火焰原子吸收分光光度计(AA-6650,岛津,日本)测定。
1.2.2细菌宏基因组16S rDNA测序
利用土壤基因组DNA提取试剂盒(FastDNA® Spin Kit for Soil)提取土壤微生物基因组DNA。总DNA用1%琼脂糖电泳进行检测,再稀释10倍后用于PCR扩增。利用Qubit2.0 DNA检测试剂盒对基因组DNA进行精确定量,以确定PCR反应应加入的DNA量。 PCR所用的引物已经融合了MiSeq测序平台的V3—V4区通用引物,引物序列见表1。

表1 测序引物及序列
PCR体系包括10×PCR buffer 5 μL、dNTP(每种10 mmol/L) 0.5 μL、基因组DNA 10 ng、Bar-PCR primer F (50 μmol/L) 0.5 μL、Primer R (50 μmol/L) 0.5 μL、Plantium Taq (5 U/μL) 0.5 μL,补充无菌水至50 μL。PCR扩增条件为:94℃预变性3 min;94℃变性30 s,45℃退火20 s、65℃延伸30 s,共5个循环;94℃变性20 s,55℃退火30 s、72℃延伸30 s,共20个循环;最后72℃延伸5 min。PCR结束后,对PCR产物进行琼脂糖电泳,用琼脂糖回收试剂盒(cat:SK8131)对DNA进行回收。回收产物用Qubit2.0 DNA检测试剂盒定量,根据测得的DNA浓度,将所有样品按照1∶1的比例进行混合,混合后充分震荡均匀。该混合样品由上海生工生物工程有限公司进行后续的样品建库与宏基因测序。
1.3 分析方法
1.3.1根际细菌多样性指标分析
将多条序列按其序列间的距离进行聚类,根据序列之间的相似性作为域值分成操作分类单元(Operational taxonomic unit, OTU),序列相似性域值设为0.97。采用uclust软件(uclust v1.1.579)对OTU进行聚类,uclust首先筛选出序列中最长的reads作为种子序列,找出所有与该序列的相似度在阈值范围内的序列,并归为一个类,而后依次执行此步骤,直到所有序列均完成聚类,每一个类作为一个OTU。
细菌Alpha多样性指标包括丰富度指数、Shannon-Wiener指数、均匀度、ACE指数、Chao1指数和覆盖度。丰富度指数通过OTU的个数来计算,香农-威纳指数衡量群落的异质性,通过公式(1)进行计算,其中Pi为第i种细菌数量占细菌总量的比值。均匀度按公式(2)计算,其中S为细菌的总物种数。
(1)
EH=H/lnS
(2)
ACE指数用以估计群落中含有的OTU数目,是生态学中估计物种总数的常用指数之一,按公式(3)计算:
(3)
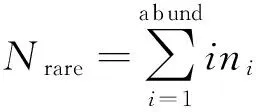
其中,ni=含有i条序列的OTU数目;Srare=含有“abund”条序列或者少于“abund”的OTU数目;Sabund=多于“abund”条序列的OTU数目;abund=“优势”OTU阈值,默认为10。
Chao1指数也是估计群落中含OTU 数目的指数。计算公式如下:
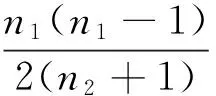
(4)
其中,SChao1=估计的OTU 数;Sobs=实际OTU 数;n1=只有一条序列的 OTU 数目;n2=只有两条序列的 OTU 数目。
覆盖率数值越高,则样本中序列没有被测出的概率越低。该指数反映了本次测序结果是否代表样本的真实情况。计算公式为:
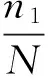
(5)
其中,n1=只含有一条序列的 OTU 的数目;N=抽样中出现的总序列数目。
细菌群落Beta多样性可以用来比较多组样本之间的差别,本研究利用Fast UniFrac软件进行主坐标分析(Principal co-ordinates analysis, PCoA)。
1.3.2基于Canoco软件的冗余分析(RDA)
利用Canoco(version4.5, Centre for Biometry, Wageningen, The Netherlands)软件分析土壤理化因子和细菌群落之间的关系。首先,对茵陈蒿根际土壤细菌群落做降趋势对应分析(Detrended correspondence analysis, DCA)。结果显示,四个排序轴中最长轴为1.03,小于3,因此本研究选择基于线性模型的冗余分析(Reundancy analysis, RDA)方法进行排序[19]。
蒙特卡罗置换检验(Monte Carlo permutation)用以检验限制性排序模型的显著性,置换次数选择默认值499次。预筛选(Forward selections)可以检验哪种环境因子对细菌群落组成有显著性的影响。根据预筛选的结果,通过偏冗余分析(partial RDA)的variation partitioning能够确定具有显著性影响的各因子的贡献率[19]。
1.3.3数据分析
使用SPSS20.0对土壤理化性质数据进行统计分析,先进行正态分布和方差齐性检验,然后做单因素方差分析。不同处理的均值在5%的显著性水平下做LSD(Least significant difference)多重比较。
2 结果与分析
2.1 土壤理化性质
采样点土壤理化性质见表2[4]。结果显示,土壤均属于酸性土,不同样点的酸度有显著差异(P<0.05),其中样方Cu1的pH最高。土壤的总铜平均浓度在7.81—25.76g/kg之间,有效铜平均浓度在1.31—5.13g/kg之间,远远超过《土壤环境质量标准(GB15618—1995)》中的三级标准限值,重金属铜污染严重。3个采样点土壤铜浓度成递增趋势,其中Cu3与其他两个采样点之间铜浓度有显著差异(P<0.05)。土壤中有机质的含量在6.40—29.35g/kg之间,3个采样点之间均有显著差异(P<0.05),总磷含量在0.22—0.53g/kg之间。总锌浓度平均在0.22—0.43g/kg之间,Cu3采样点的总锌、有效锌水平显著高于其他两个采样点。所有采样点均受到铜、锌两种重金属的复合污染。
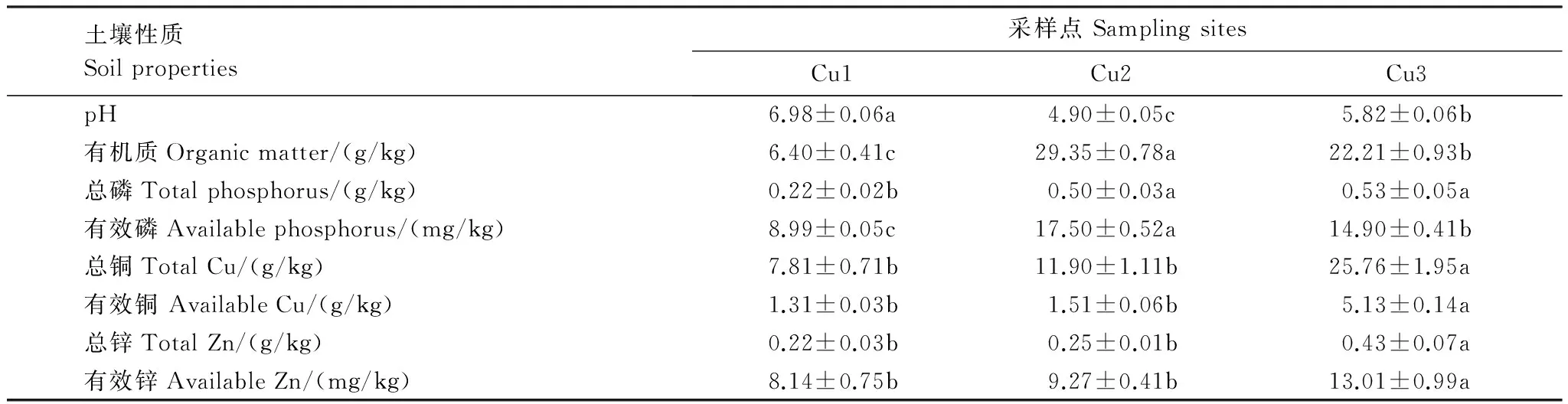
表2 土壤理化性质
表中数据为平均值±标准误差,用不同字母标记的数据表示在5%的显著性水平下有显著性差异P<0.05
图1 茵陈蒿根际土壤细菌16S rDNA基因V3—V4区琼脂凝胶电泳图Fig.1 PCR amplification targeting the V3—V4 region of bacterial 16S rDNA geneCu1、Cu2和Cu3代表W3个采样点,M 标记物GM331,Marker GM331
2.2 16S rDNA扩增产物
土壤细菌总DNA片段长度约为10000 bp左右。16S rDNA基因V3—V4区扩增产物1.5%的琼脂糖凝胶电泳检测结果见图1(Marker: GM331)。特异性引物对能够扩增出16S rDNA基因V3—V4区片段,扩增产物大约600 bp。目的片段亮度高,没有非特异性扩增,可用于后续宏基因测序。
2.3 微生物Alpha多样性指标
所有采样点测序文库的覆盖度均达到90%以上,说明绝大部分细菌的序列可以被测出,测序结果有较好的代表性。细菌丰富度指数用于衡量各采样点细菌种类数量,通过OTU的个数来计算,数量均在5700条以上,表明各采样点的细菌丰富度非常高。在铜浓度较低的Cu1采样点,其丰富度、香农威纳指数、ACE指数和Chao1指数分别为11503、7.40、40088和26476,在3个采样点中均为最大值。Cu1和Cu2两个采样点的所有多样性指数均无明显差异,与土壤铜、锌的结果相一致。但是,除覆盖度以外,两个采样点的其他所有指数均显著高于Cu3(P<0.05,表3)。
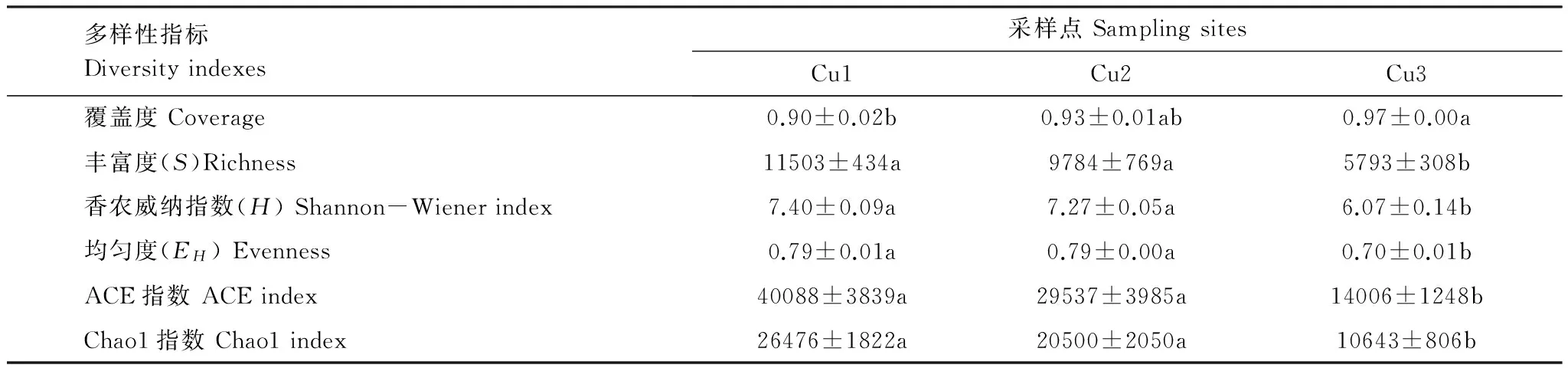
表3 土壤细菌群落多样性指标
不同字母标注的数值表示在5%的显著性水平下有显著性差异
2.4 土壤中细菌的主要类群及分布

图2 各采样点主要细菌类群相对丰度 Fig.2 The relative abundance of the major bacterial phylogenetic groups in different sampling sites
图2显示了铜矿废弃地土壤中相对丰度较高的细菌类群所占的百分比。各采样点相对丰度排名前10的优势细菌所占比例均达到95%以上,其中8个类群在3个采样点中是相同的,包括Proteobacteria(变形菌门)、Acidobacteria(酸杆菌门)、Bacteroidetes(拟杆菌门)、Gemmatimonadetes(芽单胞菌门)、Actinobacteria(放线菌门)、Verrucomicrobia(疣微菌门)、Planctomycetes(浮霉菌门)和Unclassified(未分类门)等。在采样点Cu1中,变形菌门占比高达51.63%,虽然其他两个采样点变形菌门优势度也最高,但与Cu1相比其占比明显下降(P<0.05),拟杆菌门和芽单胞菌门变化趋势与此类似。酸杆菌门在所有采样点中均排第2位,但与采样点Cu1相比,Cu2和Cu3中酸杆菌门的占比明显增加(P<0.05),放线菌门、疣微菌门、绿弯菌门(Chloroflexi)和未分类门的变化趋势与此类似,尤其是绿弯菌门,其在Cu3中的占比高达15.27%,居第3位。
2.5 细菌群落PCoA分析
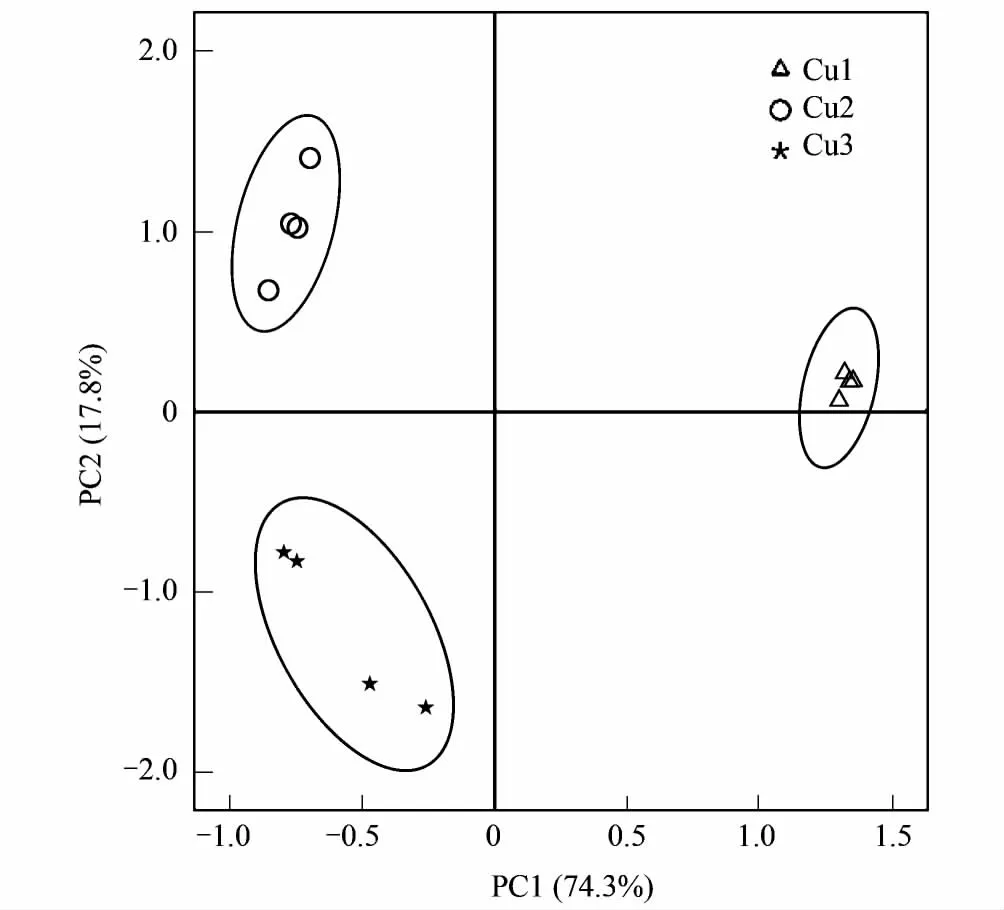
图3 各采样点细菌群落PCoA聚类 Fig.3 The PCoA analysis of bacterial communities from different sampling sites
由图3可见,3个采样点的所有4个重复均聚类于同一象限,说明组内变异相对较小,重复性较好,而群落之间位置较远,组间差异明显。PCoA分析结果表明,采样点之间细菌群落组成的变异受11个主坐标成分的控制,前5个主坐标成分的特征值均大于1%,其中前两个主坐标成分影响最大,分别能够解释74.3%和17.8%的变异,累计解释能力达92.07%。
2.6 土壤细菌群落与环境因子之间的关系
蒙特卡罗置换检验结果显示,第一典范轴和所有典范轴的P值分别为0.004(F=11.145)和0.002(F=14.720),表明该排序模型的解释变量(即土壤环境因子)可以很好地解释响应变量(即土壤细菌群落结构)的变化,且第一典范轴的影响十分显著。茵陈蒿根际土壤细菌群落与根际土壤环境因子的冗余分析结果见表4。土壤细菌群落结构与4个典范轴均具有很高的相关性(>0.969),其中RDA前两个排序轴的特征值分别为0.788和0.135,分别解释了78.8%和13.5%的细菌物种变化。本文所选的8个土壤环境因子的特征值共解释了97.5%的总方差,对茵陈蒿根际土壤细菌的物种梯度分布有十分显著的影响。
为了找到可能影响茵陈蒿根际土壤细菌群落组成的主要因子,对8个理化因子进行了预筛选。分析结果表明:总铜、总磷、pH、有效磷和有机质能显著影响茵陈蒿根际土壤细菌的群落组成(P< 0.05)。 根据预筛选的结果,对上述5个因子进行Variation partitioning分析,从而得到各因子独立影响细菌群落的强度[20]。
对茵陈蒿根际土壤细菌群落具有显著性影响的理化因子单独解释的贡献率和总贡献率结果见表5。从表中可以看出,土壤总铜对细菌群落的变化解释能力最高,为60.6%(P=0.002),其次为总磷、pH、有效磷和有机质,解释能力依次为26.8%、26.3%、25.0%和21.0%,蒙特卡罗置换检验显示其P值均达到显著水平。上述5个因子总的解释能力为93.9%,因此有65.8%的解释能力来自5个因子的重叠部分,说明因子之间对土壤细菌群落的影响存在较大相关性。

表4 土壤细菌群落冗余分析
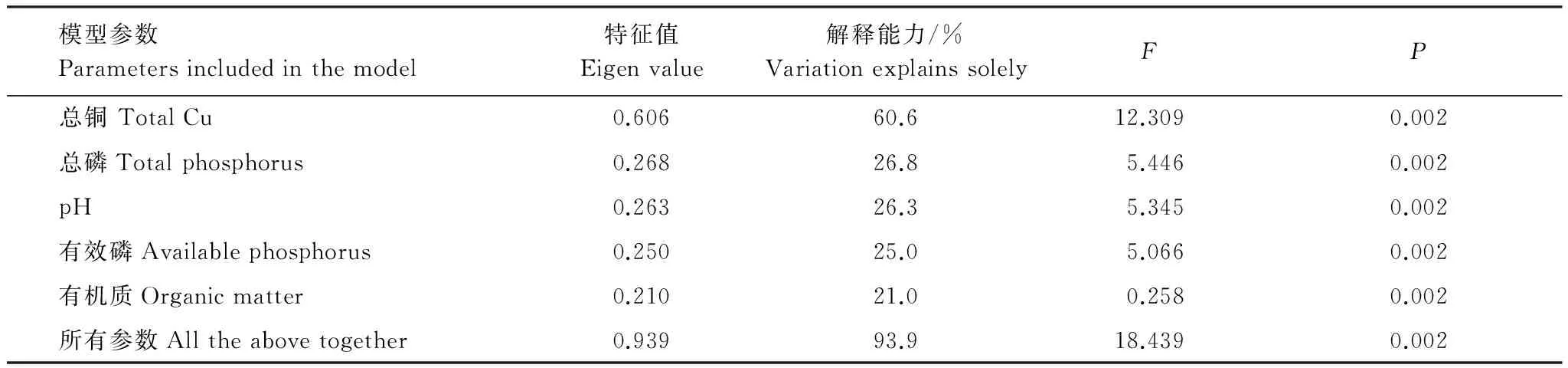
表5 土壤环境因子偏冗余分析(pRDAs)
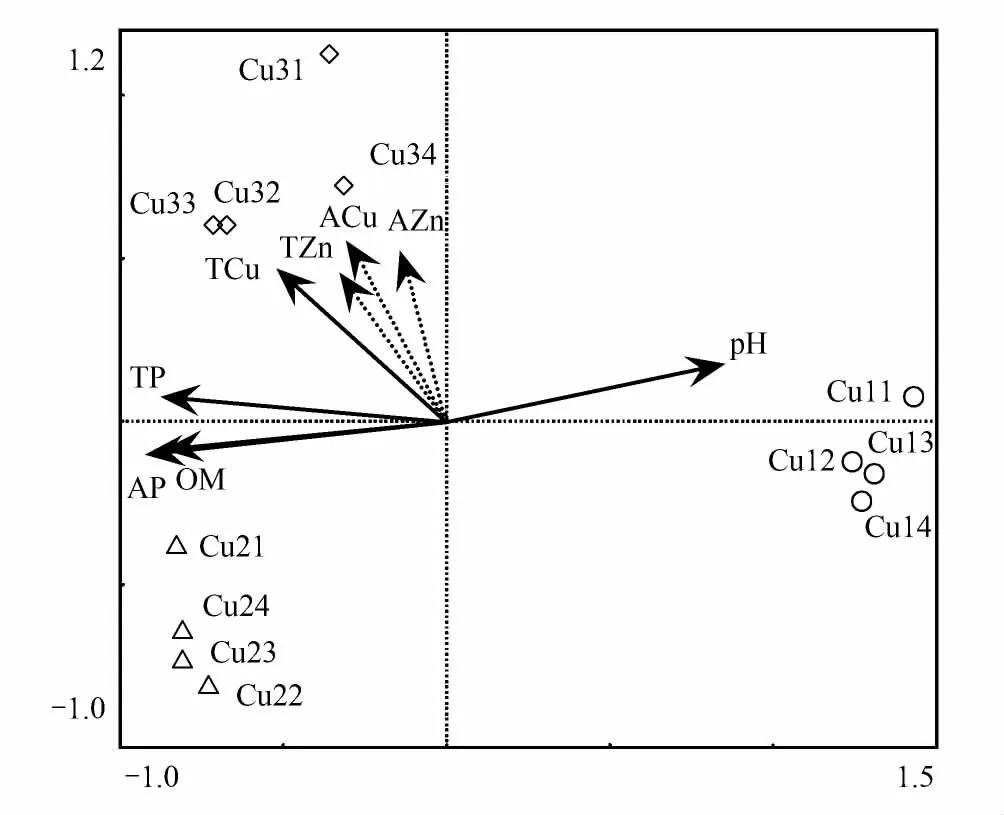
图4 采样点和土壤理化因子RDA双序图 Fig.4 The RDA biplots generated from samples and environmental variables 图中实线和实心箭头表示显著性因子,虚线和空心箭头为非显著性因子;Cu11—Cu14表示样点1的4个重复,其他样点与此相同;OM:有机质,organic matter;TP:总磷,total phosphorus;AP:有效磷,available phosphorus;TCu:总铜,total Cu;ACu:有效铜,available Cu;TZn:总锌,total Zn;AZn:有效锌,available Zn
双序图(图4)显示了各采样点的细菌群落与土壤理化因子间的关系。各采样点的4个重复均相距较近,表明采样点内部的变异相对较小,这与PCoA的结果是一致的。对采样点1影响最大的是土壤pH,其他因子与该采样点的细菌群落呈负相关。采样点2则主要受有效磷、有机质、总磷和总铜的影响,与土壤pH则呈负相关。影响采样点3的主要因子是总铜,其次是总磷、有效磷和有机质。可以看出,影响采样点2和3细菌群落的因子相似,但顺序不同,两者与采样点1之间均有较大差异。
3 讨论
3.1 茵陈蒿根际细菌群落变化特征
根际微生物在植物吸收、运输和积累营养元素、重金属的过程中发挥着重要作用,但目前对微生物群落及其功能的变化研究偏少,对上述变化产生的生态学意义的关注更显不足。 笔者前期的研究表明,随着植物群落的演替和环境污染程度的提高,丛枝菌根真菌(arbuscular mycorrhizal fungi, AMF)群落在短时间内物种组成和相对丰度均产生了明显变化,其对宿主的功能和偏好性(preference)也发生改变,从而影响植物间的相互作用,反作用于植物群落[21- 23]。本研究利用Illumina公司的MiSeq高通量测序平台对耐铜植物茵陈蒿根际的土壤细菌群落进行了宏基因组测序和分析。该技术与变性梯度凝胶电泳(denaturing gradient gel electrophoresis, DGGE)指纹图谱和传统测序方法相比,具有前处理简单、通量高、速度快、精度高、信息量大等优势,尤其对群落中的稀有种具有更高的检出能力[24- 25],已成为微生物生态学研究中的重要方法之一。本研究发现,采样点Cu1和Cu2的细菌群落多样性指标没有差异,但细菌群落的丰富度、香农威纳指数、ACE指数、Chao1指数和均匀度均显著高于Cu3。从3个采样点的土壤性质看,Cu1的有机质、总磷、有效磷等营养条件均显著低于其他两个采样点,因此物种丰富度和多样性的降低可能主要源于铜和锌对细菌产生的胁迫作用。从本次测序的覆盖度来看,Cu3却显著高于Cu1,这可能是由于Cu3中细菌种类少,多样性低,优势种优势度十分明显,非优势种所占比例很低,从而导致绝大多数细菌都能被所检出。
本次测序采用RDP classifier软件进行物种分类,共分为门、纲、目、科、属等5个层次。分类越精细其数量越大,3个采样点所检出的属均在700个以上,因此为便于分析本研究从最高分类单元进行比较。各采样点的优势细菌门优势度非常明显,排名前10的细菌门占比达95%以上,而且优势细菌门在3个采样点中绝大部分是相同的,趋同现象明显。这可能是由于铜胁迫对耐铜能力相对较弱的细菌产生了显著的选择压力,导致耐铜细菌获得了相对的优势。与此类似,通过DGGE分析该铜矿废弃地上海州香薷根际的AMF群落,结果显示AMF均来自Glomus属,表明该属具有较强的耐铜能力[22]。但是,优势细菌对铜的耐受能力仍有明显差异。变形菌门在所有采样点中优势度均最高,但在Cu2和Cu3中其优势度显著低于Cu1,拟杆菌门和芽单胞菌门同样如此;酸杆菌门、放线菌门、疣微菌门、绿弯菌门和未分类门的变化则刚好与此相反。这可能跟不同类别的细菌对环境的适应能力强弱有关,其中的主导因子应该仍是铜胁迫。值得注意的是绿弯菌门,其在Cu1中的相对丰度仅为0.54%,但在Cu2和Cu3中分别达到4.54%和15.27%,增加非常显著,意味着该菌具有极强的耐铜能力。
3.2 茵陈蒿根际细菌群落的影响因素
颜庆云等[26]发现,武汉东湖5个站点的浮游生物PCR-DGGE指纹结构的聚类结果与重金属浓度的聚类分枝关系一致,即重金属是影响浮游生物群落组成的重要因子。为了进一步探讨影响茵陈蒿根际细菌群落的环境因子,本研究做了PCoA和(偏)冗余分析。分析结果表明,各采样点内部均具有较好的重复性,这可能跟较小范围(20 m2)内的废弃地土壤具有较高的同质性有关。但是,3个采样点则明显聚成不同的群,说明细菌群落之间的差异十分明显。PCoA分析显示,细菌群落的变化与11个主坐标成分有关,但主要受前几个主坐标成分影响,其中前2个主坐标成分的解释能力达到92.07%,前8个主坐标成分可以解释99.04%的总方差。本研究所测定的8个土壤环境因子通过冗余分析投影成4个典范轴,总解释能力达到97.5%,说明茵陈蒿根际细菌群落与这8个因子具有非常密切的关系。前2个典范轴的累计解释能力达到92.3%,与PCoA前2个主坐标成分的结果接近。偏冗余分析进一步明确了对茵陈蒿根际细菌群落具有显著性影响的环境因子的贡献率。影响最大的是总铜,其单独解释能力达到60.6%,5个因子的总解释能力达到93.9%。双序图则清晰地显示了影响各个采样点细菌群落的关键因子和影响程度。采样点Cu3铜浓度最高,耐铜的酸杆菌门、放线菌门、疣微菌门、绿弯菌门和未分类门相对丰度与Cu1相比均显著增加,因此该采样点与总铜浓度呈正相关关系。采样点Cu1主要受pH影响,与总铜呈负相关,可能是因为Cu1中的变形菌门、拟杆菌门和芽单胞菌门对铜污染的敏感性相对较高,pH则可以调节铜的生物有效性,从而降低铜的胁迫作用。虽然,偏冗余分析的结果显示有效铜、总锌、有效锌对细菌群落影响不显著,但是Pearson相关分析表明,上述3个因子以及总铜与细菌群落的丰富度、多样性和均匀度均呈显著负相关(P<0.05)。 这意味着偏冗余分析主要是从细菌群落整体结构的变化而非群落的多样性进行分析的。
研究植物修复过程中根际细菌群落的变化,以及影响细菌群落结构的驱动因子,对于进一步完善植物修复技术,加速重金属污染的植物-微生物联合修复具有重要参考意义[16, 27]。
[1] 中华人民共和国环境保护部、国土资源部. 全国土壤污染状况调查公报. 北京: 环境保护部, 国土资源部, 2014: 3.
[2] 王庆仁, 崔岩山, 董艺婷. 植物修复——重金属污染土壤整治有效途径. 生态学报, 2001, 21(2):326- 331.
[3] Barillas J R V, Quinn C F, Pilon-Smits E A H. Selenium accumulation in plants-phytotechnological applications and ecological implications. International Journal of Phytoremediation, 2011, 13(S1): 166- 178.
[4] Yang R Y, Guo F Y, Zan S T, Zhou G, Wille W, Tang J J, Chen X, Weiner J. Copper tolerantElsholtziasplendensfacilitatesCommelinacommunison a copper mine spoil. Plant and Soil, 2015, 397(1/2): 201- 211.
[5] Jeong S, Moon H S, Nam K. Increased ecological risk due to the hyperaccumulation of As inPteriscreticaduring the phytoremediation of an As-contaminated site. Chemosphere, 2015, 122: 1- 7.
[6] Ma Y, Rajkumar M, Zhang C, Freitas H. Inoculation ofBrassicaoxyrrhinawith plant growth promoting bacteria for the improvement of heavy metal phytoremediation under drought conditions. Journal of Hazardous Materials, 2016, 320: 36- 44.
[7] 李韵诗, 冯冲凌, 吴晓芙, 石润. 重金属污染土壤植物修复中的微生物功能研究进展. 生态学报, 2015, 35(20): 6881- 6890.
[8] Ma Y, Oliveira R S, Freitas H, Zhang C. Biochemical and molecular mechanisms of plant-microbe-metal interactions: relevance for phytoremediation. Frontiers in Plant Science, 2016, 7: 918.
[9] Wood J L, Tang C X, Franks A E. Microbial associated plant growth and heavy metal accumulation to improve phytoextraction of contaminated soils. Soil Biology and Biochemistry, 2016, 103: 131- 137.
[10] Ullah A, Heng S, Munis M F H, Fahad S, Yang X Y. Phytoremediation of heavy metals assisted by plant growth promoting (PGP) bacteria: a review. Environmental and Experimental Botany, 2015, 117: 28- 40.
[11] Kafantaris F C A, Borrok D M. Zinc isotope fractionation during surface adsorption and intracellular incorporation by bacteria. Chemical Geology, 2014, 366: 42- 51.
[12] Gan M, Song Z B, Zhu J Y, Liu X X. Efficient bioleaching of heavy metals from contaminated sediment in batch method coupled with the assistance of heterotrophic microorganisms. Environmental Earth Sciences, 2016, 75(6): 457.
[13] Gunawan C, Teoh W Y, Marquis C P, Amal R. Cytotoxic origin of copper(II) oxide nanoparticles: comparative studies with micron-sized particles, leachate, and metal salts. ACS Nano, 2011, 5(9): 7214- 7225.
[14] Chen Y X, Wang Y P, Lin Q, Luo Y M. Effect of copper-tolerant rhizosphere bacteria on mobility of copper in soil and copper accumulation byElsholtziasplendens. Environment International, 2005, 31(6): 861- 866.
[16] Wood J L, Zhang C, Mathews E R, Tang C, Franks A E. Microbial community dynamics in the rhizosphere of a cadmium hyper-accumulator. Scientific Reports, 2016, 6: 36067.
[17] 娄晓祎, 杨如意, 郭耀广, 王兆慧, 杨慧萍, 柳建设. 铜陵市主要铜矿废弃地耐铜植物的铜积累能力比较研究//2012中国环境科学学会学术年会论文集(第三卷). 北京: 中国环境科学学会, 2012: 2631- 2634.
[18] 鲍士旦. 土壤农化分析(第三版). 北京: 中国农业出版社, 2000.
[20] Borcard D, Legendre P, Drapeau P. Partialling out the spatial component of ecological variation. Ecology, 1992, 73(3): 1045- 1055.
[21] Zhang Q, Yang R Y, Tang J J, Yang H S, Hu S J, Chen X. Positive feedback between mycorrhizal fungi and plants influences plant invasion success and resistance to invasion. PLoS One, 2010, 5(8): e12380.
[22] Yang R Y, Zan S T, Tang J J, Chen X, Zhang Q. Variation in community structure of arbuscular mycorrhizal fungi associated with a Cu tolerant plant-Elsholtziasplendens. Applied Soil Ecology, 2010, 44(3): 191- 197.
[23] 杨如意, 昝树婷, 李静, 苏楠楠, 孙雯雯, 郭富裕, 周刚. 铜胁迫对丛枝菌根真菌群落功能的影响. 应用生态学报, 2015, 26(5): 1563- 1570.
[24] 夏围围, 贾仲君. 高通量测序和DGGE分析土壤微生物群落的技术评价. 微生物学报, 2014, 54(12): 1489- 1499.
[25] 蔡言安, 李冬, 毕学军, 曾辉平, 张杰. 基于不同测序技术的生物群落结构及功能菌分析. 中国环境科学, 2016, 36(6): 1830- 1834.
[26] 颜庆云, 余育和, 冯伟松. 武汉东湖浮游生物宏基因组与环境重金属的关系. 中国环境科学, 2010, 30(S1): 52- 56.
[27] 李杰, 贺纪正, 马延和, 朱永官, 张蕾. 生物耐铜的分子机理及铜污染环境的生物联合修复. 生态学报, 2007, 27(6): 2615- 2626.
Thebacterialcommunitystructureassociatedwithacopper-tolerantplant,ArtemisiacapillariesThunb.,anditsinfluencingfactors
SHAO Zongyuan, WANG Yue, ZHANG Ju, YANG Cheng, ZHOU Gang, YANG Ruyi*
CollegeofEnvironmentalScienceandEngineering,AnhuiNormalUniversity,Wuhu241002,China
The properties and roles of indigenous heavy metal-tolerant microbes have been extensively investigated for plant growth promotion and ecological remediation at contaminated sites, but these studies seldom address the community-level features of the microbes. Three soil samples were collected from the rhizosphere of a Cu-tolerant plant,ArtemisiacapillariesThunb., grown on a Cu mine spoil. The MiSeq high-throughput sequencing technique, targeting the V3—V4 region of the bacterial 16S rDNA gene, was used to investigate the bacterial community structure and analyze the links between the bacterial community and soil environmental parameters. The results showed that sampling site Cu3 contained higher concentrations of Cu and Zn than Cu1 and Cu2 did. The Shannon-Wiener diversity index, richness, evenness, ACE index, and Chao1 index of the bacterial communities in Cu3 were all lower than those of Cu1 and Cu2, but coverage by the bacterial communities in Cu3 was higher than that of Cu1. The top 10 dominant phyla of bacteria accounted for 95% of the total relative abundance and 8 out of the top 10 dominant phyla were the same across all three sampling sites; these were Proteobacteria, Acidobacteria, Bacteroidetes, Gemmatimonadetes, Actinobacteria, Verrucomicrobia, Planctomycetes, and Unclassified. The relative abundances of Proteobacteria, Bacteroidetes, and Gemmatimonadetes were higher in Cu1 than in Cu2 and Cu3, but the opposite was true for Acidobacteria, Actinobacteria, Verrucomicrobia, Chloroflexi, and Unclassified phyla, indicating that different bacteria responded differently to Cu stress. The relative abundance of Chloroflexi, which only accounted for 0.54% of the bacterial community in Cu1, was 4.54% and 15.27% in Cu2 and Cu3, respectively. Bacterial communities were clustered into three different groups, according to principal coordinates analysis (PCoA) and redundancy analysis (RDA). The variation in bacterial communities was controlled by 11 principal coordinates, among which the first and second coordinates explained 74.3% and 14.8% of the total variance, respectively. Soil environmental parameters were closely related to the differences in bacterial community and explained 97.5% of the total variance. Total Cu, total P, pH, available P, and organic matter were the significant parameters; altogether, they accounted for 93.9% of the total variance in bacterial community. Total Cu was the most powerful factor, and explained 60.6% of the total variance independently. However, the dominant parameters differed across sampling sites. The RDA bioplots revealed that the bacterial community in Cu3, which showed the highest Cu tolerance, was positively related to total Cu. In contrast, the relatively Cu-sensitive bacterial community in Cu1 was positively correlated with pH and negatively correlated with total Cu. It is of vital importance to study how bacterial communities in the plant rhizosphere change with the environment and screen functional bacteria for plant-microbe remediation of heavy metal contamination.
heavy metal contaminated soils; phytoremediation; rhizosphere bacteria; community structure; high-throughput sequencing
国家自然科学基金资助项目(41001368); 安徽省自然科学基金资助项目(1508085SMC211); 2016年高校优秀青年人才支持计划重点项目(gxyqZD2016025)
2016- 08- 26; < class="emphasis_bold">网络出版日期
日期:2017- 07- 12
*通讯作者Corresponding author.E-mail: yangruyi@mail.ahnu.edu.cn
10.5846/stxb201608261743
邵宗圆,王悦,张菊,杨程,周刚,杨如意.耐铜植物茵陈蒿根际细菌群落结构及影响因素.生态学报,2017,37(22):7679- 7688.
Shao Z Y, Wang Y, Zhang J, Yang C, Zhou G, Yang R Y.The bacterial community structure associated with a copper-tolerant plant,ArtemisiacapillariesThunb., and its influencing factors.Acta Ecologica Sinica,2017,37(22):7679- 7688.
