基于线粒体COI基因序列的丽文蛤群体遗传多样性和遗传结构分析
2018-01-03田云方叶莹莹吴常文傅泽钦
田云方,叶莹莹,吴常文,傅泽钦
(浙江海洋大学海洋科学与技术学院,国家海洋设施养殖工程中心,浙江舟山 316022)
基于线粒体COI基因序列的丽文蛤群体遗传多样性和遗传结构分析
田云方,叶莹莹,吴常文,傅泽钦
(浙江海洋大学海洋科学与技术学院,国家海洋设施养殖工程中心,浙江舟山 316022)
为了评估丽文蛤的遗传多样性,为丽文蛤的保护与开发提供基础遗传信息,本研究利用线粒体COI基因片段序列比较分析了我国沿海丽文蛤4个地理群体(漳州、珠海、海口和北海)的遗传多样性和遗传结构。4个群体共54个样本的线粒体COI基因部分序列经处理得到长度均为623 bp的核苷酸片段,检测到19个单倍型。群体遗传多样性分析显示,4个群体的单倍型多样性为0.464~0.889,核苷酸多样性为0.000 80~0.011 04。单倍型多样性较高的是漳州群体和海口群体,北海群体单倍型多样性相对较低。核苷酸多样性中漳州群体最高,而北海群体同样显示最低,北海群体多样性较低的原因可能是样本量小。分子变异分析(AMOVA)结果显示96.20%的变异来自群体内。群体间遗传分化指数显示漳州与珠海和北海群体之间存在较大的遗传分化(FST值分别为0.162 49和0.117 31),且均达到显著水平;而漳州与海口群体之间遗传分化小且分化不显著。珠海、海口和北海群体之间遗传分化小(FST值0.070 99~0.094 30)且分化不显著。聚类分析和单倍型网络图均显示丽文蛤漳州群体与珠海和北海群体分化明显,而珠海、海口和北海群体之间分化不明显,其结果与FST值相一致。
丽文蛤;线粒体COI;遗传多样性;遗传结构
丽文蛤Meretrix lusoria属瓣鳃纲,帘蛤科,文蛤属,生活在潮间带和浅海沙质海底。它内含丰富的蛋白质,具有高营养、易养殖的特点,是我国沿海滩涂养殖的主要品种之一。丽文蛤外观与文蛤极为相似,庄启谦[1]提出文蛤属在我国仅有3种,即文蛤M.meretrix、丽文蛤和斧文蛤M.lamarckii。关于文蛤和斧文蛤的研究有很多,但是对于丽文蛤的研究仅限于形态学[2]及外缘凝集素[3]和贝壳采集[4]的相关研究。而关于丽文蛤遗传差异的研究仅有陈爱辉等[5]基于线粒体COI基因序列的文蛤属(软体动物门:帘蛤科)系统发育关系。针对于丽文蛤线粒体DNA标记研究丽文蛤的遗传结构的文章,未有报道。
由于近年来过度捕捞和比较严重的海洋污染,文蛤属各物种的群体多样性面临极大挑战,由于丽文蛤在外形上与文蛤存在着诸多相似,一直被忽视,游离在研究之外,尚未从分子水平加以研究分析。潘宝平等[6]首先利用分子生物学手段对丽文蛤和文蛤、斧文蛤的亲缘性进行了研究。陈爱辉等[7]基于线粒体COI基因序列的文蛤属的系统发育研究。TORRI,et al[8]中国,韩国和印度的丽文蛤的研究,标明是关于丽文蛤的形态及遗传特征的研究,但是文章内容着重研究形态学及丽文蛤的分类,并没有采用分子手段对丽文蛤群体遗传多样性进行研究。而国内尚未有采用分子手段对丽文蛤的群体多样性进行研究的报道。
细胞色素C氧化酶亚基I(COI)是线粒体DNA中一个重要基因,基因变异性较大、进化速率比较快,在采用分子手段研究贝类[9-10]和物种群体的遗传多样性及结构分析[11-13]等方向应用广泛。本文采用线粒体COI基因进行标记,对丽文蛤四个不同沿海地理种群的分子遗传学研究分析,为我国丽文蛤种质资源的研究和丽文蛤及文蛤属物种多样性研究提供理论依据。
1 材料与方法
1.1 样品采集和DNA提取
本研究所用丽文蛤分别来源于4个地理群体,为广西北海(BH)、海南海口(HK)、广东珠海(ZH)、福建漳州(ZZ)如图1。每个地点随机采样活体解剖取闭壳肌(表1),保存于无水乙醇中,用于DNA提取。采用改良盐析法[14]提取总DNA,利用1%的琼脂糖电泳进行检测,无菌超纯水稀释至50 ng/μL备用。
1.2 线粒体COI序列扩增
丽文蛤线粒体COI扩增引物来源于通用引物LCO1490和HCO2198[15],COI-F 5'-GGTCAACAAATCATAAAGATATTGG-3'COI-R 5'-TAAACTTCAGGGTGACCAAAAAATCA-3'PCR反应体系为 25.0 μL,内含 Taq 酶 mix 12.5 μL,正、反向引物各 1.0 μL(10 μmol/L),模板 DNA 1.0 μL,ddH2O 9.5 μL。PCR 反应程序为 94 ℃预变性为 2 min,94 ℃变性时间为30 s,52℃退火时间为30 s,72℃延伸时间为1 min,35个循环反应后,72 ℃延伸时间为10 min。PCR产物经1%~2%琼脂糖凝胶电泳检测后,PCR产物最后送杭州擎科(杭州擎科梓熙生物技术有限公司)进行双向测序。
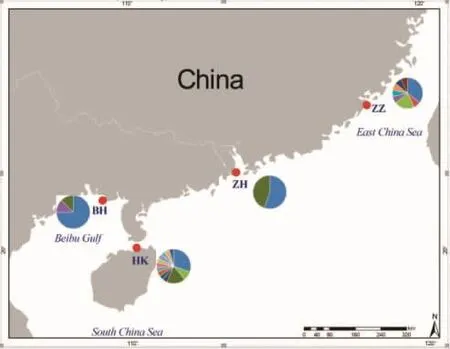
图1 本研究丽文蛤地理群体采集点地图Fig.1 Collection locations of M.lusoria geographical populations in this study

表1 丽文蛤4个群体单倍型多样性及核苷酸多样性Tab.1 Haplotype diversity and nucleotide diversity of four populations of M.lusoria
1.3 数据统计与分析
实验测序全部采用双向测序,为了确认样品分类的准确性,序列结果均在NCBI(https://www.ncbi.nlm.nih.gov/)上进行Blast对比,统计好单倍型并上传NCBI(https://www.ncbi.nlm.nih.gov/)数据库,并获得登录号(表2)。利用Mega版本5.0[16]对获得的COI序列进行整理编辑,结合人工对编辑结果进行矫正。利用DNASP[17]统计单倍型,计算每个实验群体的单倍型多样性(h)和核苷酸多样性(π)。利用Arlequin 3.5[18]软件计算群体间及群体内的遗传分化指数(FST),并进行P检验,通过10 000次重复置换检验(Permutation test)分析其显著性。同时分析群体内和群体间的分子方差(Analysis of Molecular Variance,AMOVA),以10 000次重复抽样检验其显著性,并进行Fu`s Fs检验。利用Network 5.0.0.1以核苷酸进化模型计算各单倍型之间的遗传距离并构建单倍型网络图,利用 Mega 5.0[15]以Kimura 2-parameter模型建立4个群体的Unweighted Pair Group Method with Arithmatic Mean(UPGMA)单倍型系统进化树,抽样次数设置为1 000次。
2 结果
2.1 群体内遗传变异
对4个丽文蛤地理群体的DNA进行PCR扩增,获得所需的COI基因片段,经BLAST比对分析,确认所得片段为丽文蛤COI序列,经Mega同源排序,去除部分端部序列,获得54条623 bp的COI基因片段。序列组成显示平均 A、T、G、C 碱基含量分别为 A:11.43%、T:41.54%、C:23.92%、G:23.11%。其中 A+T:52.97%、C+G:47.03%。A+T含量大于G+C的含量,具有明显的碱基组成偏倚性。共检测出单倍型19个(表2)(登录号MF285209-MF285227),其中H1被漳州、珠海、海口和北海群体共享,H3被漳州和海口群体共享,Hap9被珠海和海口群体共享。H2、H5、H6、H7、H8为漳州群体特有单倍型,H10-19为海口群体特有单倍型。
丽文蛤4个地理群体的遗传学参数见表2,平均单倍型及核苷酸多样性处于中等水平(h=0.851,π=0.040 01)。单倍型多样性最高的是漳州群体(h=0.945)和海口群体(h=0.917),北海群体单倍型多样性相对较低(h=0.750)。核苷酸多样性中漳州群体最高(π=0.179 7),而北海群体核苷酸多样性显示最低(π=0.014 7)。北海群体和珠海群体的单倍型多样性和核苷酸多样性偏低,这可能是由于样本量比较小,但是我们不排除样品本身的原因造成珠海和北海本身丽文蛤的遗传多样性低于其他地区。
2.2 群体遗传结构研究
用MEGA5.0构建4个群体单倍型系统发育树如图2,系统发育树可以看出,单倍型9以下没有珠海和海口群体出现。漳州和海口群体大部分群体都聚集在下方,且没有明显分化。用Network5.0.1.1构建单倍型网络图如图3,结果显示单倍型网络图右侧没有珠海和北海群体出现,这表明漳州与珠海和北海群体发生了显著分化。这与系统发育树和FST值结果一致。
丽文蛤4个群体COI序列的分子变异分析(AMOVA)结果见表4,群体间遗传变异占5.38%,群体内遗传变异占94.60%,群体内的遗传变异大于群体间,群体间分化系数P值小于0.05,达到显著水平,表明群体间发生显著的遗传分化(P<0.05)。

表2 单倍型序列比对Tab.2 Sequence comparison of haplotypes
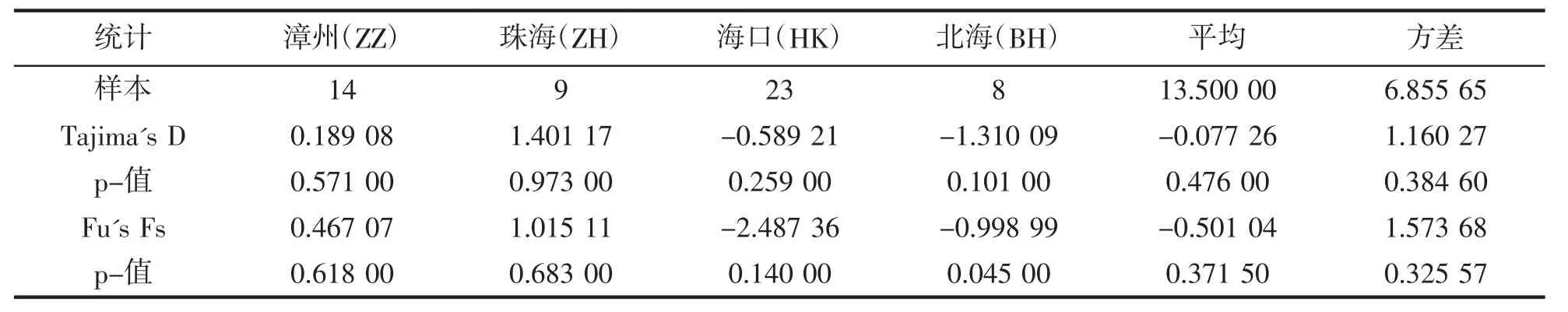
表3 丽文蛤4个群体的COI序列选择中性检验Tab.3 Selective neutrality test analysis of 4 populations of M.lusoria based on COI sequences

图2 丽文蛤4个群体的单倍型UPGMA系统进化树Fig.2 Haplotype UPGMA tree of 4 populations of M.lusiria
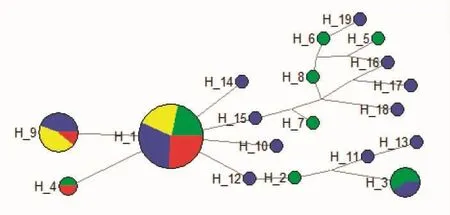
图3 丽文蛤4个群体的单倍型网络图Fig.3 Haplotype network of 4 populations of M.lusiria

表4 丽文蛤COI序列遗传差异的分子方差分析(AMOVA)Tab.4 Analysis of molecular variance(AMOVA)of COI gene of M.lusiria
丽文蛤群体间的FST显著性检测见表5,两两之间的差异数值介于0.018 16~0.162 49之间,其中珠海群体和漳州群体(FST=0.162 49)(P<0.05)、北海群体和漳州群体(FST=0.117 31)(P<0.05),说明珠海群体和漳州群体,北海群体和漳州群体发生了显著分化。从地理位置来看,珠海所在地理位置位于珠江三角洲,海洋生物类易于堆积。而北海所在位置与漳州之间隔有陆地,这可能就是阻挡基因交流的重要原因。漳州与海口之间没有阻隔,斧文蛤群体以及幼体均可以随海流和潮流四处移动,基因间交流较大,分化不明显。中性检验结果显示,平均Fu`s Fs值为-0.501 04(P>0.05)(表3),Fu`s Fs值未达到显著水平,表明丽文蛤未偏离中性选择,丽文蛤群体没有经历过群体扩张。
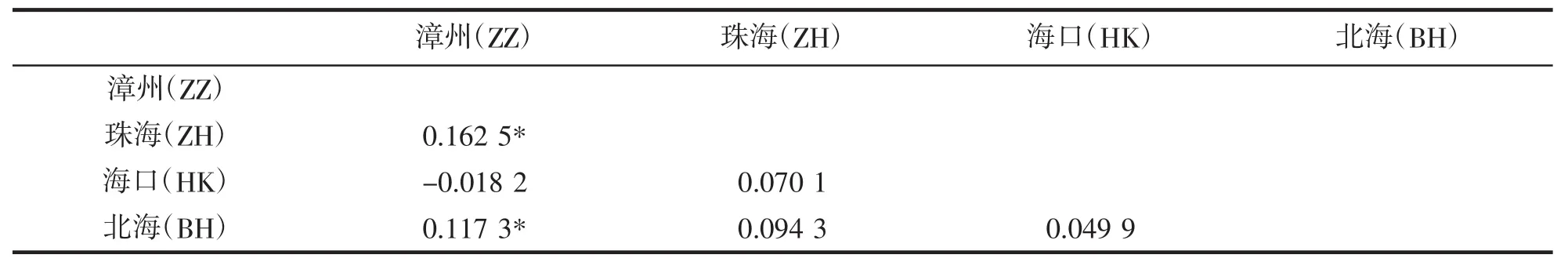
表5 基于线粒体COI序列的丽文蛤群体遗传分化Tab.5 Estimates of population genetic differentiation based on COI genefor the M.lusiria
3 讨论
线粒体COI基因具有高度的保守性,一般多可以用于物种的多样性比较。本研究检测到丽文蛤线粒体COI序列的单倍型多样性为0.790,核苷酸多样性为0.007 27,与我国其他海水贝类[19-21]相比,遗传多样性不高。这可能与我们实验样本量不足引起的,但也有可能我们实验所采集的样本中有效群体小、群体之间亲本性别比例不平衡等[22]造成,从而导致丽文蛤群体得COI基因序列遗传多样性偏低。从单倍型网络图来看,各群体单倍型2~10个不等。本实验研究样品来源于我国主要的文蛤养殖海域,由于丽文蛤与文蛤在形态学上难以鉴定,文蛤各群体的养殖规模大,我们也可以说是丽文蛤养殖规模比较大。养殖群体的群体遗传多样性偏低,放殖到野生环境中与野生群体想混合生殖,导致野生群体的遗传多样性整体偏低。
群体遗传中的遗传分化系数是衡量物种分化的重要指标,本文4个群体丽文蛤群体间的FST为0.053 8(P<0.05),表明群体间的相似性不大,群体间发生了显著分化,而群体内的遗传变异达到0.946 2(P<0.05),群体内分化显著。其中群体间珠海群体和漳州群体(FST=0.162 49)(P<0.05)、北海群体和漳州群体(FST=0.117 31)(P<0.05),漳州与珠海和北海群体间分化显著。某些双壳类必要时可凭借贝壳的急剧开合和外套膜触手的作用在海中进行蝶式游泳。大部分水生贝类营底栖生活,或在水底匍匐、爬行,或在底质中挖穴隐居,或附着在其他外物上生活。所以贝类会随着海流和潮流进行移动[23],本研究中的四个地理位置中珠海位于珠江三角洲,海洋生物易于堆积,而不易于迁移。北海与漳州之间被海峡阻隔,基因交流不大。相对来说珠海、海口与北海之间相距较近,基因交流的机会较大,所以群体间没有明显的遗传分化。漳州与海口之间虽然相距甚远但是地理位置间没有阻隔,随着海流与潮流,基因交流的可能性较大,因此群体间分化不明显。实验所采用的样品为野生样品,流动性较大,这可能与野生群体的异地养殖和增殖放流有关。李宏俊等[24]也认为贝类的养殖可能对当地野生群体的遗传结构造成影响。单倍型系统发育树显示,漳州群体和珠海和北海群体发生了显著分化,系统发育树H9以下没有珠海和北海群体出现,单倍型网络图显示右侧没有珠海和北海群体出现,表明珠海群体和北海群体与漳州群体发生明显的分化,这与地理环境和海流有关[23],也可能物种本身经过长时间的自身遗传进化所引起的。
我们衡量物种进化潜力的依据是分析物种的遗传多样性和遗传结构,通常情况下,遗传多样性的高低代表物种适应环境的能力。本研究表明丽文蛤群体在经历长时间的进化,已经产生了明显的分化,这表明丽文蛤自身的适应能力在逐步提升。但是由于近年来海洋污染的加剧,海洋生物面临极大的挑战,很多物种面临灭绝的危险。文蛤属仅有三种贝类,文蛤及斧文蛤的研究明显对于丽文蛤,丽文蛤的分子研究也一直游离于研究之外,由于丽文蛤与文蛤在外形上极为相似,不易分辨,所以丽文蛤的养殖及野生群体的总体数量并不是很清楚,这很不利于丽文蛤种资资源的保护与开发。我们的研究结果将为丽文蛤的深入研究及其开发保护提供帮助。
4 小结
本文采用4个野生丽文蛤为样本对丽文蛤的遗传差异进行线粒体COI分子研究,结果显示群体间发生了明显的分化,群体内分化较为明显,这可能与文蛤群体的野生异地养殖和增值放流有关,但不排除丽文蛤本身的基因特点。丽文蛤作为文蛤属其中一种,研究丽文蛤的遗传差异对文蛤属的整体物种多样性具有重要意义。目前关于丽文蛤的研究相对较少,这对其本身的发展及保护不利。丽文蛤和文蛤相类似,比较容易养殖,而且味道鲜美,所以今后丽文蛤的养殖和野生驯养以及野生群体和养殖群体的差异性比较等都是值得研究的课题。
[1]庄启谦.中国动物志软体动物门双壳纲帘蛤科[M].北京:科学出版社,2001:229-236.
[2]陈爱华,吴杨平,姚国兴,等.丽文蛤与文蛤4个地理群体的形态差异分析[J].海洋渔业,2010,30(2):125-131
[3]朱越雄,曹广力.丽文蛤血清中外源凝集素的凝集性能[J].海洋科学,2000(10):55-56.
[4]小池裕子,李慧冬,王 青.丽文蛤在推断绳文时期贝类采集季节中的作用[J].南方文物,2008(3):130-133.
[5]陈爱辉,李朝霞,封功能.基于线粒体 COI基因序列的文蛤属(软体动物门:帘蛤科)系统发育关系[J].动物学研究,2009,30(3):233-239.
[6]潘宝平,吴 琪,张素萍,等.文蛤属(Meretrix)16SrRNA基因及ITS1序列的系统学分析[J].海洋与湖沼,2006,37(4):342-347.
[7]陈爱辉,李朝霞,封功能.基于线粒体 COI基因序列的文蛤属(软体动物门:帘蛤科)系统发育关系[J].动物学研究,2009,30(3):233-239.
[8]TORII H S,SATO S C,et al.The comparison of shell morphology and geneticrelationship between Meretrix lusoria and M.petechialis in Japan and Korea.Plankton and Benthos Research[J].Plankton&Benthos Research,2010(5):231-241.
[9]程汉良,夏德全,吴婷婷,等.6种帘蛤科贝类及4个地理种群文蛤线粒体COI基因片段序列分析[J].海洋学报,2007,29(5):109-116.
[10]程汉良,彭永兴,董志国,等.基于线粒体细胞色素c氧化酶亚基I基因序列的帘蛤科贝类分子系统发育研究[J].生态学报,2013,33(9):2 744-2 753.
[11]牛东红,李家乐,沈和定,等.缢蛏六群体线粒体DNA-COI基因序列变异及群体遗传结构分析[J].海洋学报,2008,30(3):109-116.
[12]沈玉帮,张俊彬,冯冰冰,等.基于线粒体COI序列分析对紫贻贝群体遗传多样性的研究分析[J].海洋通报,2011,30(4):435-440.
[13]ZHAN A B,PEREPELIZIN P V,GHABOOLI S,et al.Scale-dependent post-establishment spread and genetic diversity in an invading mollusc in South America[J].Diversity and Distributions,2012,18(10):1 042-1 055.
[14]ALJANABI S M,MARTINEZ I.Universal and rapid salt-extraction of high quality genomic DNA for PCR-based techniques[J].Nucleic acids Res,1997(25):4 692-4 693.
[15]FOLMER O,BLACK M,HOEH W,et al.DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates[J].Molecular Marine Biology Biotechnology,1994,3(5):294-299.
[16]TAMURA K,DUDLEY J,NEI M,et al.MEGA 4:molecular evolutionary genetics analysis(MEGA)software version 4.0[J].
Molecular Biology and Evolution,2007,24(8):1 596-1 599.
[17]ROZAS J,SÁNCHEA-DELBARRIO J C,MESSEGUER X,et al.DNA polymorphism analyses by the coalescentand other methods[J].Bioinformatics,2003,19(18):2 496-2 497.
[18]EXCOFFIER L,LISCHER H E L.Arlequin suite ver 3.5:a new series of programs to perform population genetics analyses under Linux and Windows[J].Molecular Ecology Resources,2010,10(3):564-567.
[19]王 超,陈爱华,曹 奕,等.6个不同海域文蛤地理群体的亲缘关系分析[J].海洋渔业,2016,38(3):262-272.
[20]郑文娟,朱世华,沈锡权,等基于线粒体COI基因序列探讨泥蚶的遗传分化[J].动物学研究,2009,30(1):17-23.
[21]MAO Y L,GAO T X,YANAGIMOTO T,et al.Molecular phylogeography of Ruditapes philippinarum in the Northwestern Pacific Ocean based on COI gene[J].Journal of Experimental Marine Biology and Ecology,2011,407(2):171-181.
[22]AVISE J C,ARNOLD J,Ball R M,et al.Intraspecificphylogeography:the mitochondrial DNA bridge between population genetics and systematics[J].Annual Review of Ecology and Systematics,1987,18:489-522.
[23]马 超.黑潮对东中国海主要流系的影响[D].青岛:中国海洋大学,2009.
[24]李宏俊,张晶晶,袁秀堂,等.利用线粒体COI和微卫星标记分析文蛤7个地理群体的遗传变异[J].生态学报,2016,36(2):499-507.
Genetic Diversity and Genetic Structure of Meretrix lusoria Population based on Mitochondrial COI Gene Sequences
TIAN Yun-fang,YE Ying-ying,WU Chang-wen,et al
(School of Ocean Science and Technology of Zhejiang Ocean University,National Engineering Research Center For Marine Aquaculture,Zhoushan 316022,China)
In order to evaluate the genetic diversity of Meretrix lusoria,make a contribution to protect and develope the M.lusoria.In this study,the genetic diversity of four geographical populations(Zhangzhou,Zhuhai,Haihou and Beihai)of M.lusoriawas calculated based on the mitochondrial cytochrome c oxidase subunit I(COI)gene sequences.A 623 bp nucleotide fragment was obtained in the 54 individuals in all 4 populations and 19 haplotypes were detected in them.Population genetic diversity analysis showed that the haplotype diversity of each population was 0.464-0.889.Zhangzhou shows the highest haplotype diversity in the four populations,and the haplotype diversity of Beihai is relatively low.This may be caused by a small sample size.Molecular variation analysis(AMOVA)results showed 96.20%variation from within the population.The genetic differentiation indexes between Zhangzhou/Zhuhai and Zhangzhou/Beihai populations showed a large genetic differentiation (FSTvalues is 0.162 49 and 0.117 31,respectively),and reached a significant level.The genetic differentiation between Zhangzhou and Haikou was not significant.The genetic differentiation between Zhuhai/Beihai and Haikou/Beihai populations were not significant.Cluster analysis and haplotype network map showed that the differentiation between Zhangzhou and Zhangzhou populations was significant,while the differentiation among Zhuhai,Haikou and Beihai populations werenot obvious,and the results were consistent with the FSTvalues.
Meretrix lusoria;MT COI;genetic diversity;genetic structure
Q959.215+.4
A
2096-4730(2017)04-0320-06
2017-05-10
浙江海洋大学“水产”省一流学科开放课题(20160002)
田云方(1990-),女,河南上蔡人,硕士研究生,研究方向:海洋生物学.E-mail:xiaotian081215@126.com
叶莹莹,研究方向:分子遗传育种.E-mail:Yeyingying5559@163.com
