激动素对AlCl3和D-半乳糖联合诱导的AD小鼠模型大脑病理变化和Aβ表达的影响
2017-12-26魏云鹏欧阳五庆
刘 丹,魏云鹏,欧阳五庆
(西北农林科技大学动物医学院,陕西杨陵 712100)
激动素对AlCl3和D-半乳糖联合诱导的AD小鼠模型大脑病理变化和Aβ表达的影响
刘 丹,魏云鹏,欧阳五庆*
(西北农林科技大学动物医学院,陕西杨陵 712100)
旨在研究激动素对AD小鼠模型的神经保护作用。首先用AlCl3和D-半乳糖联合处理制备AD小鼠模型,然后用不同浓度的激动素进行干预处理,再通过HE染色对大脑海马CA3区进行病理学观察,并对大脑皮层和海马部位β淀粉样蛋白(β amyloid protein,Aβ)进行免疫组化染色。结果表明,AlCl3和D-半乳糖处理过的小鼠其海马CA3区锥体细胞发生明显的病理变化,而激动素的干预减轻了这一病变,且表现出剂量依赖性。同时,AlCl3和D-半乳糖处理过的小鼠与空白组小鼠相比,大脑皮层和海马区Aβ1-42分布均显著增加,但激动素干预能够使Aβ1-42的表达和分布明显减少,且显示出剂量依赖性的特点。这些结果说明激动素对AD小鼠模型具有一定的正向干预作用,表明激动素可能具有抗AD的潜力。
激动素;三氯化铝;D-半乳糖;病理变化;Aβ
阿尔茨海默病(Alzheimer′s disease,AD)是一种神经退行性疾病,发病率随年龄增长而急剧上升,其能够剥夺老年患者的自主生活能力,并严重危害老年人的身心健康。AD的典型病理学特征包括大脑皮层和海马区细胞外出现大量由Aβ沉积形成的老年斑(senile plaque,SP)[1],神经元细胞内出现大量由高度磷酸化tau蛋白形成的神经纤维缠结(neurofibrillary tangle,NFT)[2]和明显的神经元丢失[3]。尽管AD的发病机制多样,但已有研究表明铝暴露和氧化应激是AD发病的重要原因[5-7]。Al是地壳中含量最丰富的金属元素,但其对大脑有着非常明显的神经毒性[8]。铝(Al)会引起神经元细胞外Aβ沉积并诱发活性氧(reactive oxygen species,ROS)产生,进而引起神经元氧化应激和损伤[9]。D-半乳糖是一种体内的还原糖,当其在体内含量大大高于正常水平时,它可被半乳糖氧化酶氧化为不可被进一步代谢的醛类,其大量蓄积于脑组织中,从而引发神经细胞氧化应激和衰老凋亡[10]。大量研究表明,三氯化铝(AlCl3)和D-半乳糖联合处理动物,尤其是啮齿动物,能够显著损伤其学习和记忆能力,并能诱导大量的Aβ1-42在大脑皮层和海马部位表达和积累[11-12],且联合使用能增强两者的神经毒性效应[13]。目前,联合应用AlCl3和D-半乳糖制备AD小鼠模型已得到国内外的广泛认可。
激动素属于植物细胞分裂素家族成员,是腺嘌呤的衍生物。研究发现,激动素可以调节植物细胞分裂和分化,同时能够增强植物细胞抗衰老及抗氧化能力[14-16]。此外,其他相关研究表明,激动素同样能够在动物细胞中发挥抗衰老和抗氧化损伤的作用。同时,有报道称激动素能够保护大鼠大脑和体外培养的星形胶质细胞免于D-半乳糖诱导的氧化应激造成的损伤[16-17]。但激动素是否对AD小鼠模型具有神经保护作用还未见报道。本研究利用AlCl3和D-半乳糖联合制备小鼠AD模型,同时用不同浓度的激动素进行干预,然后通过观察大脑病理变化及Aβ分布水平来研究激动素对AD小鼠模型的神经保护作用。
1 材料与方法
1.1 材料
1.1.1 实验动物 健康成年雌性昆明小鼠,90只,体重18 g~22 g,均未孕,购于第四军医大学。
1.1.2 主要试剂 激动素(纯度≥97%)为Sigma公司产品(K0753);D-半乳糖为Solarbio公司产品(D8310);兔抗鼠Aβ1-42一抗为Abcam公司产品(ab201060);HRP山羊抗兔二抗为Solarbio公司产品(SE134)。
1.1.3 主要仪器 病理切片机为上海徕卡仪器有限公司产品(RM2016);正置光学显微镜为日本尼康公司产品(NIKON ECLIPSE CI)。
1.2 方法
1.2.1 动物分组与处理 将90只小鼠随机平均分为6组,分别为空白组、模型组、激动素低剂量干预组、激动素中剂量干预组、激动素高剂量干预组、激动素组。模型组和低、中、高激动素干预组每日饲喂含有AlCl3饮用水,每只小鼠每天摄入AlCl3的剂量为100 mg/kg,同时于颈背部注射200 mg/kg 的D-半乳糖。另外对低、中、高激动素干预组分别进行激动素混悬液灌胃,激动素混悬液的灌胃剂量分别为10 mg/kg、20 mg/kg和40 mg/kg;激动素组每日饲喂正常饮用水,颈背部注射等量的生理盐水,同时用40 mg/kg剂量的激动素混悬液灌胃一次;空白组每日饲喂正常饮用水,颈背部注射等量的生理盐水,同时灌胃等量的蒸馏水。所有操作连续进行90天。
1.2.2 脑组织固定和取材 以400 mg/kg剂量的巴比妥钠对小鼠实施腹腔麻醉,再用剪刀和镊子打开胸腔,在右心耳处剪开一个小口,将装有生理盐水的注射器针头插入左心室,进针不超过0.5 cm。将生理盐水缓缓推进小鼠心脏,右心耳流出的液体逐渐变淡至无色,然后将注射器拔离针头,针头不要移动,快速将装有40 mL/L多聚甲醛的去针头注射器插入,先快后慢推进小鼠体内,可看到小鼠四肢紧张,尾部卷曲。待小鼠心脏完全停止跳动,撤去注射器,用弯头剪刀小心打开并剥离颅骨,再用弯镊伸入颅底离断颅底神经,取出全脑,放于40 mL/L多聚甲醛中固定24 h以上。
1.2.3 病理组织学观察 将固定好的全脑经梯度乙醇脱水,二甲苯透明,浸蜡,包埋,切片,HE染色,对海马区进行观察拍照并分析。
1.2.4 免疫组织化学染色 将1.2.3制备的石蜡切片脱蜡,抗原修复,内源性过氧化物酶阻断,BSA封闭,一抗孵育,二抗孵育,DAB染色,细胞核复染及脱水封片,对皮质和海马区进行观察拍照并分析。
2 结果
2.1 小鼠大脑海马CA3区病理组织学变化结果
光学显微镜下观察表明,空白组小鼠大脑海马CA3区的锥体细胞没有明显的形态变化以及病理学变化,细胞整体排列密集整齐、胞体完整、核仁明显;模型组的锥体细胞排列错乱、细胞核固缩、细胞深染、形状不规则;激动素低剂量干预组的锥体细胞病理学变化较模型组略有改善,正常形态的锥体细胞数量略有增多,异常锥体细胞数量略有减少;激动素中剂量干预组相比低剂量干预组出现更多正常的锥体细胞及更少的异常锥体细胞;而激动素高剂量干预组相比中剂量干预组出现更多正常的锥体细胞及更少的异常锥体细胞;激动素组的锥体细胞与空白组相比,没有明显的形态变化和病理学变化。
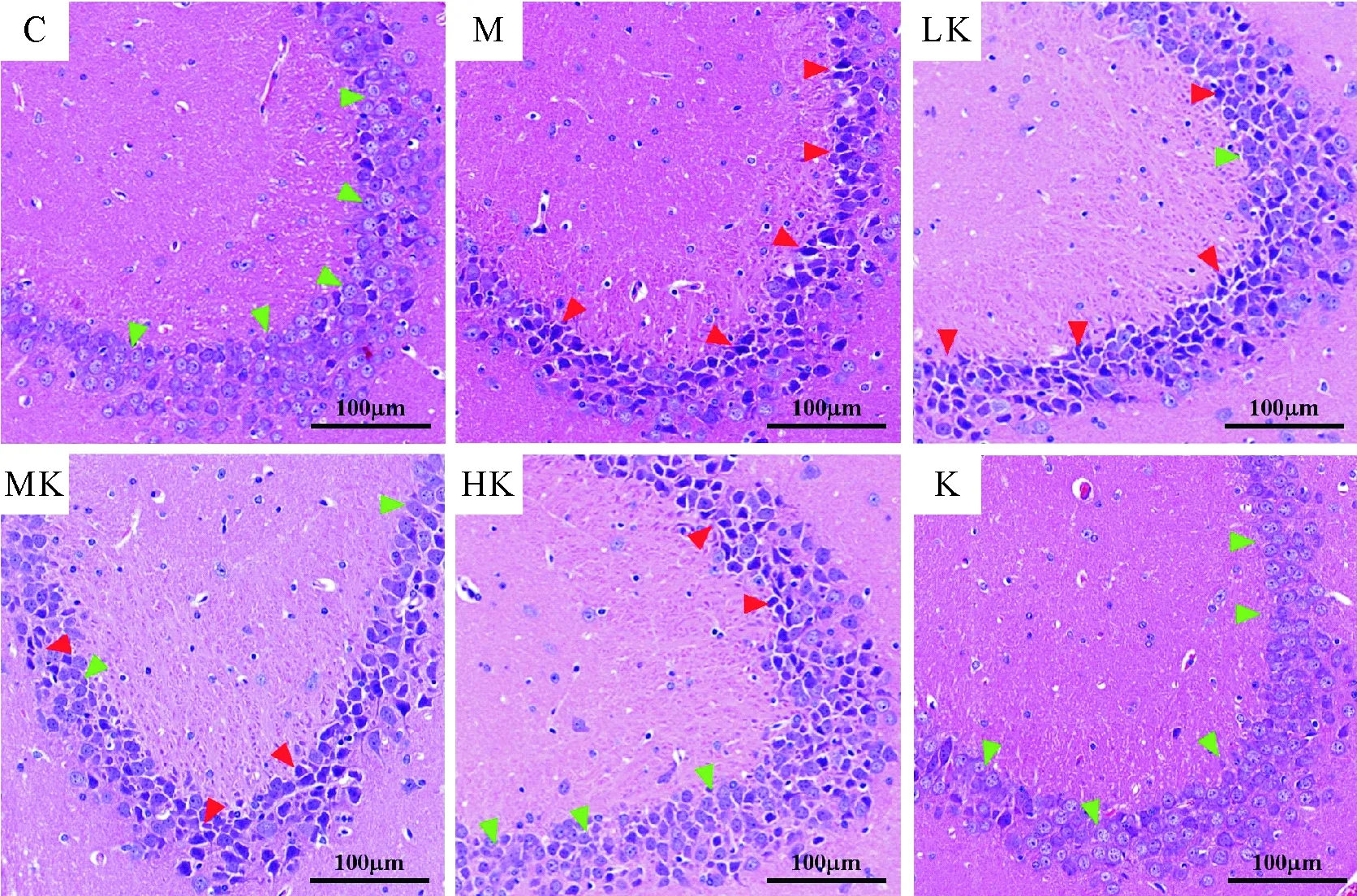
C.空白组;M.模型组;LK.激动素低剂量干预组;MK.激动素中剂量干预组;HK.激动素高剂量干预组;K.激动素组。绿色箭头所指的是正常的锥体细胞,而红色箭头所指的是非正常锥体细胞
C.Control group; M.Model group; LK.Low dose kinetin group; MK.Middle dose kinetin group; HK.High dose kinetin group; K.Kinetin group.The black arrowhead points normal pyramidal cells and red arrowhead points distorted pyramidal cells
图1各组小鼠大脑海马CA3区HE染色显微图像
Fig.1 Photomicrograph of hippocampus CA3 region from each experimental group
2.2 免疫组织化学染色结果
免疫组织化学染色结果显示,空白组小鼠大脑皮层中染成棕色Aβ1-42斑块很少;模型组小鼠大脑皮层区神经元细胞周围存在大量染成深棕色的Aβ1-42斑块,同时神经元间隙也弥散性地广泛存在染成浅棕色的Aβ1-42;激动素低剂量干预组的神经元细胞周围也存在较多染成棕色的Aβ1-42斑块,但染色深度相比于模型组较浅,神经元细胞间隙弥散分布的Aβ1-42也相对减少;激动素中剂量干预组神经元周围和神经元间隙的Aβ1-42分布相比激动素低剂量干预组进一步降低;而激动素高剂量干预组神经元周围和神经元间隙的Aβ1-42分布和激动素中剂量干预组相比更低;激动素组小鼠大脑皮层中的Aβ1-42分布和空白对照组相比差别不大。
此外,空白组小鼠海马区锥体细胞周围被染色的Aβ1-42斑块很少;模型组小鼠海马区锥体细胞周围大量存在被染成浅棕色的Aβ1-42斑块,细胞间隙也存在广泛且弥散的Aβ1-42分布;激动素低剂量干预组相比模型组小鼠,海马区锥体细胞周围和细胞间隙的Aβ1-42分布有所下降;激动素中剂量干预组相比激动素低剂量干预组,海马区锥体细胞周围和细胞间隙Aβ1-42的分布进一步减少;激动素高剂量干预组相比激动素低剂量中预组,海马区锥体细胞周围和细胞间隙的Aβ1-42分布更少;激动素组海马区的锥体细胞周围和细胞间隙的Aβ1-42分布和空白对照组相比差别不大。

A.Aβ1-42在大脑皮层中的分布;B.Aβ1-42在大脑海马区的分布;C.空白组;M.模型组;LK.激动素低剂量干预组;MK.激动素中剂量干预组;HK.激动素高剂量干预组;K.激动素组
A. The distributions of Aβ1-42in cortex ;B.The distribution of Aβ1-42in hippocampus;C.Control group; M.Model group; LK.Low dose kinetin group; MK.Middle dose kinetin group; HK.High dose kinetin group; K.Kinetin treatment alone
图2小鼠大脑Aβ1-42免疫组化结果(200×)
Fig.2 Immunohistochemical staining results of Aβ1-42in brain tissues of each group(200×)
3 讨论
大量研究表明,铝元素具有显著的神经毒性,是已知多种神经退行性疾病的高风险致病因子[18]。首先,铝元素的促氧化能力在ROS介导的神经变性损伤中起着非常重要的作用。铝离子可以和超氧阴离子结合形成一种铝过氧化物[19],诱导线粒体产生ROS,引起氧化应激。线粒体既是ROS的主要来源,也最易遭受其攻击。线粒体膜和蛋白的损伤会产生更多的ROS,损伤线粒体DNA,激活大脑细胞凋亡信号途径,引起神经元凋亡[20]。其次,长期接触铝元素会引起大脑ATP合成速率降低,同时升高ATP水解速率[21],从而干扰大脑的能量代谢过程。再次,在大脑中,铝易与富含磷酸的核酸结合,影响遗传信息的表达,从而产生基因毒性[6-8]。此外,铝会引起细胞内蛋白和肽段沉积,而沉积的Aβ会形成老年斑并进一步产生Aβ诱导的神经毒性[9]。铝除了具有上述神经毒性,还能容易地穿过血脑屏障[18],从而大大增强了其神经毒性。D-半乳糖是一种体内的还原糖,正常情况下,其会被迅速转化为葡萄糖。当其高于正常水平时,它可被半乳糖氧化酶氧化为醛类。大量未代谢的半乳糖以及其代谢物聚集在脑组织中,引发细胞渗透压升高、细胞内ROS升高、细胞脆性增加及细胞损伤,进而导致脑功能的退化,包括认知损伤和Aβ沉积[10,22-24]。
大脑中的海马区是进行学习、记忆和空间导航的最重要部位,其从解剖学上可划分为若干个重要的区域,其中最重要的是齿状回、CA3区和CA1区[25]。其中CA3区的锥体细胞可接受来自三方面的刺激,包括齿状回颗粒细胞伸出的苔状纤维、嗅皮质浅层星型角质细胞的穿通轴突以及CA3区锥体细胞相互伸出的回返性旁支[26]。由于CA3区有着复杂的神经网络,所以其在大脑的记忆功能中扮演着重要的角色[25]。本试验中,AlCl3和D-半乳糖处理过的小鼠海马CA3区发生明显的退行性病变,例如椎体细胞排列错乱、细胞核固缩、细胞形态不规则。这些变化说明AlCl3和D-半乳糖确实具有显著的神经毒性,能够对海马区细胞造成实质性的损伤并造成显著的病理变化,这与之前的报道一致[11,27]。然而,激动素的干预减轻了这一病变,且表现出剂量依赖性,表明激动素能减轻AlCl3和D-半乳糖联合诱导的神经毒性。
Aβ是由β和γ分泌酶逐步水解β淀粉样前体蛋白(β-amyloid precursor protein,APP)所形成的短肽[1],其主要类型包括Aβ1-40和Aβ1-42,且Aβ1-42被认为比Aβ1-40具有更强的神经毒性[4]。Aβ能刺激谷氨酸受体引发兴奋性中毒,这在Aβ致病级联反应中起着重要的作用[28]。Aβ能抑制中间神经元的活性,而异常的神经网活动可能会引发恶性循环,造成更多Aβ聚集[29]。Aβ神经毒性还可能与其能够引发氧化应激有关。在AD患者和转基因小鼠模型中,Aβ能与乙醇脱氢酶在线粒体中发生反应,产生更多的ROS,使线粒体功能的丧失,最终引起细胞凋亡[30-31]。ROS能通过激活各种信号通路,改变APP代谢进程,使Aβ异常产生和积累[9]。另外,Aβ能与糖基化终末产物受体、清除剂受体和丝氨酸蛋白酶抑制剂复合酶受体结合,直接或间接激活小胶质细胞和星型细胞,释放细胞毒性物质、自由基、细胞因子等,引发脑部局部炎性反应,造成炎症病变[5]。本试验中,AlCl3和D-半乳糖处理组小鼠大脑相比空白组,其皮质和海马区Aβ1-42均显著增加,说明AlCl3和D-半乳糖处理确实能够增加小鼠大脑Aβ1-42的表达量,这与此前的报道相一致。激动素干预后Aβ1-42的表达和分布明显减少,且显示出剂量依赖性的特点。这说明激动素能抑制AlCl3和D-半乳糖诱导的Aβ1-42过量表达,其根本原因可能是激动素抑制了铝的神经毒性。已有研究表明,激动素能干扰铝在动物体内的吸收,其能减慢胃肠黏膜对铝的吸收,同时减弱尿毒症小鼠对血浆中铝的吸收。这是因为铝的吸收经由细胞旁路途径,而旁路吸收途径会被激动素阻断[17]。这可能是激动素抑制铝神经毒性的可能机制之一。
综上所述,激动素能够改善AlCl3和D-半乳糖联合处理造成的小鼠海马CA3区的病理变化,同时能够抑制AlCl3和D-半乳糖联合诱导的小鼠大脑皮层和海马区Aβ1-42的过量表达,说明激动素对AlCl3和D-半乳糖联合诱导的AD小鼠模型具有一定的正向干预作用,说明激动素可能具有抗AD的潜力。
[1] Bertram L,Lill C M,Tanzi R E.The genetics of Alzheimer disease:back to the future[J].Neuron,2010,68(2):270-281.
[2] Huang Y, Mucke L.Alzheimer mechanisms and therapeutic strategies[J].Cell,2012,148(6):1204-1222.
[3] Palop J J, Mucke L.Epilepsy and cognitive impairments in Alzheimer disease[J].Arch Neurol,2009,66(4):435-440.
[4] Tamagno E,Guglielmotto M,Monteleone D,et al.Amyloid-beta production:major link between oxidative stress and BACE1[J].Neurotox Res,2012,22(3):208-219.
[5] Bhattacharjee S,Zhao Y, Hill J M,et al.Aluminum and its potential contribution to Alzheimer's disease (AD)[J].Frontiers Aging Neurosci,2014,6(62).doi: 10.3389/fnagi.2014.00062.
[6] Lukiw W J,Kruck T P, Crapper M D.Aluminium and the nucleus of nerve cells[J].Lancet,1989,333(8641):781-781.
[7] Baydar T,Papp A,Aydin A,et al.Accumulation of aluminum in rat brain:does it lead to behavioral and electrophysiological changes[J]. Biol Trace Elem Res,2003,92(3):231-244.
[8] David S,Shoemaker M,Haley B E.Abnormal properties of creatine kinase in Alzheimer's disease brain:correlation of reduced enzyme activity and active site photolabeling with aberrant cytosol-membrane partitioning[J].Brain Res Mol Brain Res,1998,54(2):276-287.
[9] Exley C.The aluminium-amyloid cascade hypothesis and Alzheimer's disease[J].Subcell Biochem,2005,38:225-234.
[10] Cui X,Zuo P, Zhang Q,et al.Chronic systemic D-galactose exposure induces memory loss,neurodegeneration,and oxidative damage in mice:protective effects of R-alpha-lipoic acid[J].J Neurosci Res,2006,84(3):647-654.
[11] 覃广乐,李 昱.D-半乳糖、氯化铝中毒阿尔茨海默病模型小鼠脑组织Aβ蓄积原因探讨[J].山东医药,2013(30):30-32.
[12] Yang W,Shi L, Chen L,et al.Protective effects of perindopril on d-galactose and aluminum trichloride induced neurotoxicity via the apoptosis of mitochondria-mediated intrinsic pathway in the hippocampus of mice[J].Brain Res Bull,2014,109:46-53.
[13] Yang H,Qu Z, Zhang J,et al.Ferulic acid ameliorates memory impairment in d-galactose-induced aging mouse model[J].Int J Food Sci Nutr,2016,67(7):806-817.
[14] Kimura T,Doi K.Depigmentation and rejuvenation effects of kinetin on the aged skin of hairless descendants of Mexican hairless dogs[J].Rejuvenation Res,2004,7(1):32-39.
[15] Olsen A,Siboska G E,Clark B F,et al.N(6)-Furfuryladenine,kinetin,protects against Fenton reaction-mediated oxidative damage to DNA[J].Biochem Biophys Res Commun,1999,265(2):499-502.
[16] Berge U,Kristensen P,Rattan S I.Kinetin-induced differentiation of normal human keratinocytes undergoing aging in vitro[J].Ann N Y Acad Sci,2006,1067:332-336.
[17] Barciszewski J,Massino F, Clark B F.Kinetin A multiactive molecule[J].Int J Biol Macromol [J],2007,40(3):182-192.
[18] Bhattacharjee S,Zhao Y,Hill J M,et al.Aluminum and its potential contribution to Alzheimer's disease (AD)[J].Frontiers Aging Neurosci,2014, 6:62.
[19] Exley C.The pro-oxidant activity of aluminum[J].Free Radic Biol Med,2004,36(3):380-387.
[20] Ricci C,Pastukh V,Leonard J,et al.Mitochondrial DNA damage triggers mitochondrial-superoxide generation and apoptosis[J].Am J Physiol Cell Physiol,2008,294(2):413-422.
[21] Kumar V,Bal A,Gill K D.Impairment of mitochondrial energy metabolism in different regions of rat brain following chronic exposure to aluminium[J].Brain Res,2008,1232:94-103.
[22] Zhang X L,An L J,Bao Y M,et al.d-galactose administration induces memory loss and energy metabolism disturbance in mice:protective effects of catalpol[J].Food Chem Toxicol,2008,46(8):2888-2894.
[23] Lei Y,Fu W,Chen J,et al.Neuroprotective effects of abacopterin E fromAbacopterispenangianaagainst oxidative stress-induced neurotoxicity[J].J Ethnopharmacol,2011,134(2):275-280.
[24] Liu C M,Ma J Q,Lou Y.Chronic administration of troxerutin protects mouse kidney against D-galactose-induced oxidative DNA damage[J].Food Chem Toxicol,2010,48(10):2809-2817.
[25] Schwarting R K,Busse S.Behavioral facilitation after hippocampal lesion:A review[J].Behav Brain Res,2017,317:401-414.
[26] Ishizuka N,Weber J,Amaral D G.Organization of intrahippocampal projections originating from CA3 pyramidal cells in the rat[J].J Comp Neurol,1990,295(4):580-623.
[27] 罗焕敏,肖 飞.D-半乳糖和三氯化铝诱导小鼠产生类阿尔茨海默病变[J].中国药理学与毒理学杂志,2004(1):22-26.
[28] Millan S M,Heyn S N,Das D,et al.Neurobiological elements of cognitive dysfunction in down syndrome:exploring the role of APP[J].Biol Psychiatry,2012,71(5):403-409.
[29] Bero A W,Yan P,Roh J H,et al.Neuronal activity regulates the regional vulnerability to amyloid-beta deposition[J].Nat Neurosci,2011,14(6):750-756.
[30] Lustbader J W,Cirilli M,Lin C,et al.ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease[J].Science,2004,304(5669):448-452.
[31] Takuma K,Yao J,Huang J,et al.ABAD enhances Abeta-induced cell stress via mitochondrial dysfunction[J].FASEB J,2005,19(6):597-598.
EffectsofKinetinonPathologicalLesionsandAβExpressioninBrainofADMouseModelInducedbyAlCl3andD-galactoseCombinationTreatment
LIU Dan,WEI Yun-peng,OUYANG Wu-qing
(CollegeofVeterinaryMedicine,NorthwestA&FUniversity,Yangling,Shaanxi,712100,China)
The aim of this study was to investigate the neuroprotective effects of kinetin on AD mouse models.The AD mouse models were prepared by AlCl3and D-galactose,and at the same time were treated with different concentrations of kinetin.The hippocampal CA3 region was observed by HE staining.Immunohistochemical staining of β amyloid protein (Aβ) in the cortex and hippocampus was also performed.The results showed that the pyramidal cells in the hippocampal CA3 region of the mice treated with AlCl3and D-galactose had a significant pathological change,but the intervention of kinetin reduced the lesion.At the same time,the distribution of Aβ1-42in cortex and hippocampus was significantly increased compared with the blank group,but that in kinetin intervention group was significantly decreased.All the results showd kinetin dose-dependent characteristics.These results indicated that kinetin has some positive effects on AD mouse models,and may has the potential to fight AD.
kinetin; AlCl3; D-galactose; pathological lesion; Aβ
2017-04-21
陕西省重大科技创新专项基金(K332020916)
刘 丹(1992-),女,陕西西安人,硕士研究生,主要从事激动素药理学研究。*
S852.2
A
1007-5038(2017)12-0068-05
