氮杂环卡宾催化硝基甲烷与查尔酮的Michael反应
2017-12-13邢芬陈磊冯泽南理阳过杜广芬何林
邢芬,陈磊,冯泽南,理阳过,杜广芬,何林*
(石河子大学化学化工学院/新疆兵团化工绿色重点实验室,新疆 石河子832003)
氮杂环卡宾催化硝基甲烷与查尔酮的Michael反应
邢芬,陈磊,冯泽南,理阳过,杜广芬,何林*
(石河子大学化学化工学院/新疆兵团化工绿色重点实验室,新疆 石河子832003)
氮杂环卡宾作为一类重要的小分子催化剂,近年来在有机合成领域得到了广泛的应用。本文研究了氮杂环卡宾作为Brφnsted碱催化硝基甲烷与查尔酮的Michael加成反应,发展了氮杂环卡宾催化构筑γ-硝基羰基化物的有效方法。实验结果表明:以17 mol%稳定的氮杂环卡宾IPr作为催化剂,以二氯甲烷为溶剂,各种取代基的查尔酮与硝基甲烷可以在室温下顺利反应,以26%-99%的产率得到目标产物。该反应具有效率高、无金属污染、环境友好等优点。
氮杂环卡宾;Michael反应;Brφnsted碱;γ-硝基羰基化物;有机催化
氮杂环卡宾(NHCs),作为一类重要的小分子催化剂,在不对称合成领域得到了广泛的研究[1-2]。由于氮杂环卡宾中碳原子最外层只有6个电子而没有到达8电子稳定结构,使其具有不稳定性,卡宾一般以中间体的形式存在于有机反应中。1991年Arduengo等[3]制备了第1例稳定的氮杂环卡宾1,3-二-(1-金刚烷基)咪唑-2-碳烯。氮杂环卡宾作为小分子催化剂,具有催化效率高、不对称诱导效果优异、反应条件温和、无金属污染等优点,这类有机小分子催化剂在过去十年得到快速的发展,在不对称合成、天然产物全合成、药物合成等领域都获得广泛应用。根据基本骨架中杂原子的不同,氮杂环卡宾主要分为噻唑、咪唑、饱和的咪唑烷、三唑四种基本类型。基于氮杂环卡宾极其优越的亲核性能,利用对醛羰基的极性翻转策略,化学家发展了氮杂环卡宾催化的不对称的安息香缩合反应[4]、Setter反应[5]、不饱和醛的延伸极性翻转反应[6]、官能化醛[7]的转化等一系列重要反应;同时,氮杂环卡宾还被成功的应用于烯酮的不对称环加成反应[8]、惰性酯的活化[9],单电子氧化反应[10]等不对称催化领域。但是以上这些重要的反应都是基于氮杂环卡宾的路易斯碱性,而基于这类有机催化的Brφnsted碱性的研究才刚刚起步。Nolan和Hedrick[11]小组最早发现,氮杂环卡宾可以高效催化酯与醇之间的酯交换反应。机理研究表明,在反应中氮杂环卡宾是作为Brφnsted碱活化醇羟基,从而催化反应进行。
Coquerel[12]研究了1,3-二羰基化合物分子间和分子内的carba-Michael反应,该反应得到高立体选择性的螺环化合物。随后,Scheidt小组[13]实现了氮杂环卡宾催化醇与α,β-不饱和酮的oxo-Michael反应,发展了合成β-烷氧基酮的一种新方法;Scheidt[14]还尝试了手性氮杂环卡宾催化的不对称醇与α,β-不饱酮的oxo-Michael反应,虽然只得到了11%的值,但证实了基于氮杂环卡宾Brφnsted碱的催化反应可以获得手性诱导效果。在此基础上,不同的研究小组分别实现了氮杂环卡宾催化的carba、sulfa、phospha-Michael加成反应。但这类反应一直未获得手性突破。直到2014年Huang[15]首次实现了手性氮杂环卡宾催化的高选择性不对称carba-Michael加成反应,在极其精细的反应调控下,1,3-二羰基化合物可与活泼的硝基烯烃发生不对称共轭加成反应,以高达99%的ee值得到双羰基化加成产物。在大量实验的基础上,Huang等提出了氮杂环卡宾非共价键催化模型,认为六氟异丙醇作为一个氢键连接体与功能化的NHC作用,从而创造出一个微观环境来活化底物同时有利于不对称的识别。以此模型为基础,Huang课题组进一步实现了氮杂环卡宾催化的不对称aza-Michael反应和不对称sulfa-Michael加成反应[16]。
近年来,我们课题组一直从事氮杂环卡宾催化研究,发展了氮杂环卡宾催化的vinylous-Mukaiyamaaldol反应、vinylous-Mukaiyama-Michael反应[17]、氰甲基化反应等多类亲硅活化反应[18],近2年又利用氮杂环卡宾的Brφnsted碱性,发展了当归酯的立体选择性 vinylogous-Michael反应[19]、硫醇的 sulfa-Michael反应[20]、酰胺化反应[21-22]等重要反应。在这些研究工作的基础上,我们利用氮杂环卡宾独特的Brφnsted碱性,进一步实现了硝基甲烷与查尔酮的Michael反应,发展催化合成γ-硝基羰基化合物的有效方法。本文以氮杂环卡宾为催化剂,以硝基甲烷与查尔酮为模型反应,实现了分子间C-C键的构筑,得到了相应的Michael加成反应产物,并进行了条件的优化,考察了反应的适应性,并提出了催化机理。
1 实验部分
1.1 实验仪器与试剂
1H NMR(400 Hz),用TMS作为内标;采用Bruker Avance III HD 400 MHz型核磁共振仪,CDCl3为氘代试剂;所有溶剂严格按照溶剂处理手册标准进行处理;薄层色谱板为青岛海洋化工GF 254硅胶板;柱层析硅胶为青岛海洋化工厂生产(300-400目);查尔酮按照文献步骤合成[23],其他化学试剂均为分析纯或化学纯,购买于阿拉丁化学试剂公司。
1.2 实验步骤和化合物的结构表征
1.2.1 氮杂环卡宾催化Michael加成反应的通用步骤
反应管在红外干燥箱下干燥30 min后,抽换氮气3次,氮气保护下,向10 mL干燥的反应管中加入查尔酮(0.2 mmol,41.6 mg),并加入无水四氢呋喃(1 mL)搅拌溶解,然后加入硝基甲烷(0.3 mmol,18.3 mg),随后再加入催化剂 IPr(0.034 mmol,6.7 mg)。反应混合物在室温下反应12 h直至原料消失(TLC监测)。反应完成后,粗产物通过柱色谱分离即可得到纯净的目标产物(硅胶,PE-EtOAc)。
1.2.2 目标化合物结构与表征
(3a)4-Nitro-1,3-diphenylbutan-1-one:78.9 mg;97%;白色固体;mp:99.3℃-99.5℃;1H NMR(400 MHz,CDCl3)δ8.02–7.84(m,2H),7.62–7.58(m,1H),7.51–7.43(m,2H),7.43–7.23(m,5H),4.86(dd,J=12.5,6.6 Hz,1H),4.72(dd,J=12.5,8.0Hz,1H),4.34 – 4.15(m,1H),3.58–3.37(m,2H)。13CNMR(100 MHz,CDCl3)δ 196.8,139.1,136.3,133.5,129.0,128.7,128.0,127.8,127.4,79.5,41.5,39.3。
(3b)4-Nitro-1-phenyl-3-(p-tolyl)butan-1-one:82.8 mg 95.3%;白色固体;mp:100.3℃-102.4℃;1H NMR(400 MHz,CDCl3)δ7.98–7.85(m,2H),7.62–7.51(m,1H),7.50–7.40(m,2H),7.15(m,4H),4.80(dd,J=12.4,6.7 Hz,1H),4.65(dd,J=12.4,8.0 Hz,1H),4.34–4.15(m,1H),3.55–3.31(m,2H),2.30(s,3H).13CNMR(100 MHz,CDCl3)δ196.9,137.5,136.4,136.0,133.5,129.7,128.0,127.2,79,7,41.6,38.9,21.6。
(3c)3-(3-Bromophenyl)-4-nitro-1-phenylbutan-1-one:99.4mg;95.3%;白色固体;mp:103.0℃-103.5℃;1H NMR(400 MHz,CDCl3)δ7.91–7.65(m,2H),7.64–7.55(m,2H),7.36–7.30(m,2H),7.30–7.23(m,3H),4.81(dd,J=12.5,6.8 Hz,1H),4.68(dd,J=12.5,7.7 Hz,1H),4.21 (p,J=7.1 Hz,1H),3.50-3.32(m,2H);13CNMR(100 MHz,CDCl3)δ196.4,138.7,138.0,136.4,131.1,130.3,129.1,128.8,127.9,127.5。
(3d)1-(4-Bromophenyl)-4-nitro-3-phenylbutan-1-one:99.4mg;51.5%;白色固体;mp:154.8℃-155.1℃;1H NMR(400 MHz,CDCl3)δ7.80–7.74(m,2H),7.62–7.57(m,2H),7.37–7.30(m,2H),7.30–7.24(m,3H),4.81 (dd,J=12.5,6.8 Hz,1H),4.68(dd,J=12.5,7.7 Hz,1H),4.24–4.17(m,1H),3.48–3.34(m,2H);13CNMR(100 MHz,CDCl3)δ195.8,138.8,135.0,132.0,129.5,129.5,129.1,128.8,127.9,127.4,79.4,41.4。
(3e)5-Nitro-4-phenylpentan-2-one:16.2mg;26.1%;白色固体;mp:126.1℃-126.5℃;1H NMR(400 MHz,CDCl3)δ7.38–7.30(m,2H),7.30–7.24(m,1H),7.24–7.19(m,2H),4.69(dd,J=12.3,6.9 Hz,1H),4.60(dd,J=12.3,7.7 Hz,1H),4.01(m,1H),2.92(d,J=7.0 Hz,2H),2.12(s,3H)。13CNMR(100 MHz,CDCl3)δ205.3,138.8,129.0,127.9,127.3,79,46.1,39.0,30.4。
(3f)3-(Naphthalen-2-yl)-4-nitro-1-phenylbutan-1-one:90.9 mg;95%;白色固体;mp:97.5℃-98.5℃;1H NMR(400 MHz,CDCl3) δ 7.93(m,2H),7.89–7.75 (m,3H),7.62–7.52(m,1H),7.51–7.37(m,5H),4.92 (dd,J=12.5,6.6 Hz,1H),4.79(dd,J=12.5,8.0 Hz,1H),4.48–4.34(m,1H),3.62–3.49(m,2H)。13CNMR(100 MHz,CDCl3)δ196.7,136.4,136.3,133.6,133.4,132.8,129.0,128.7,128.0,127.6,126.5,126.4,126.2,125.1,79.5,41.5,39.4。
(3g)1-(3-Methoxyphenyl)-4-nitro-3-phenylbutan-1-one:72.6mg;84.7%,白色固体;mp:114.1℃-116.2 ℃;1H NMR (400 MHz,CDCl3)δ7.49(m,1H),7.43(dt,J=6.1,3.1 Hz,1H),7.37–7.30(m,3H),7.30–7.23(m,3H),7.14–7.07(m,1H),4.82(dd,J=12.5,6.7 Hz,1H),4.68(dd,J=12.5,7.9 Hz,1H),4.26–4.17(m,1H),3.83(s,3H),3.51–3.35(m,2H)。13CNMR(100 MHz,CDCl3)δ196.6,159.9,139.1,137.7,129.7,129.0,127.8,127.4,12 0.6,120.0,112.2,79.5,55.4,41.6,39.3。
(3h)3-(Furan-2-yl)-4-nitro-1-phenylbutan-1-one:72.7 mg;93.6%;白色固体;mp:76.0℃-76.5℃;1 H NMR(400 MHz,CDCl3)δ7.94(dt,J=8.5,1.6 Hz,2H),7.64–7.55(m,1H),7.51–7.43(m,2H),7.20(dt,J=10.0,5.0 Hz,1H),7.00–6.90(m,2H),4.85(dd,J=12.6,6.3 Hz,1H),4.71(dd,J=12.6,7.6 Hz,1H),4.61 – 4.49(m,1H),3.68–3.30(m,2H)。13CNMR(100 MHz,CDCl3)δ196.4,141.8,136.2,133.7,128.7,128.0,127.1,125.5,124.7,79.8,42.3,34.7。
(3i)3-(2-Chlorophenyl)-1-(4-methoxyphenyl)-4-nitrob-utan-1-one:88.9 mg;97.9%;无色液体;1H NMR (400 MHz,CDCl3)δ7.97–7.87(m,2H),7.57(m,1H),7.44(dd,J=13.7,6.3 Hz,2H),7.42–7.37(m,1H),7.32–7.26(m,1H),7.26–7.17(m,2H),4.95–4.78(m,2H),4.69(p,J=6.8 Hz,1H),3.66–3.42(m,2H)。13CNMR(100 MHz,CDCl3)δ196.7,136.2,130.4,129.0,128.7,128.0,127.4,77.4,77.1,76.7,39.9,36.1。
(3j)1-(3-Bromophenyl)-4-nitro-3-phenylbutan-1-one:89.7 mg;86%;白色固体;mp:85.3℃-85.9℃;1H NMR (400 MHz,CDCl3)δ8.03(t,J=1.7 Hz,1H),7.83(ddd,J=7.8,1.6,1.1 Hz,1H),7.70(ddd,J=8.0,2.0,1.0 Hz,1H),7.37–7.31(m,3H),7.30–7.23(m,3H),4.81(dd,J=12.5,6.9 Hz,1H),4.69(dd,J=12.5,7.7 Hz,1H),4.21(p,J=7.0 Hz,1H),3.50–3.36(m,2H)。13CNMR(100 MHz,CDCl3)δ195.4,138.8,138.0,136.4,131.1,130.3,129.1,128.0,127.4,126.5。
(3k)1-(3-Bromophenyl)-4-nitro-3-phenylbutan-1-one:113 mg;98.7%;白色固体;mp:101.8℃-103.2℃;1 H NMR(400 MHz,CDCl3)δ7.82–7.76(m,2H),7.63–7.57(m,2H),7.45–7.37(m,1H),7.30–7.18(m,3H),4.92– 4.76(m,2H),4.73 – 4.59(m,1H),3.63 – 3.36(m,2 H)。13CNMR(100 MHz,CDCl3)δ195.7,136.0,134.9,133.7,132.1,130.5,129.5,129.1,128.9,127.4,39.8,36.0。
(3m)3-(2-Bhlorophenyl)-1-(4-methoxyphenyl)-4-nitrob-utan-1-one:98.9 mg;99%;白色固体;mp:112.3 ℃-113.7 ℃;1H NMR(400 MHz,CDCl3)δ7.97–7.87(m,2H),7.42–7.37(m,1H),7.31–7.17(m,3H),6.97–6.87 (m,2H),4.93–4.79(m,2H),4.67(p,J=6.8 Hz,1H),3.85 (s,3H),3.56 – 3.39(m,2H)。13CNMR(100 MHz,CDCl3) δ 195.1,136.4,133.8,130.4,130.3,129.3,128.9,127.3,113.9,55.5,39.5,36.2。
(3n)3-(Naphthalen-1-yl)-4-nitro-1-phenylbutan-1-one:93.7 mg;98%;白色固体;mp:103.2℃-103.1℃;1H NMR (400 MHz,CDCl3)δ8.22(d,J=8.5 Hz,1H),7.94–7.84(m,3H),7.77(dt,J=7.3,3.7 Hz,1H),7.62–7.47(m,3H),7.46–7.36(m,4H),5.17(p,J=6.9 Hz,1H),5.00–4.75(m,2H),3.70–3.52(m,2H)。13C NMR(100 MHz,CDCl3)δ196.1,136.3,135.1,134.2,133.6,131.0,129.2,128.7,128.4,128.0,126.9,126.1,125.3,12 2.4,78.8,41.4。
2 结果与分析
2.1 NHC催化Michael反应
反应式如下:

2.2 NHC催化Michael反应的条件优化
首先,以硝基甲烷与不带取代基的查尔酮反应作为模型反应a,考察不同的氮杂环卡宾催化剂、溶剂等反应参数对Michael反应产率的影响,结果见表1。

表1 氮杂环卡宾催化剂、溶剂、催化剂用量对Michael反应的影响aTab.1 Effect of NHCs,base,solvent and additive on Micheal reactiona
首先,以THF作为溶剂,考察催化剂对反应的影响,分别考察咪唑、三唑、噻唑三类氮杂环卡宾对反应的影响,实验结果表明:
(1)噻唑7与三唑卡宾8催化剂不催化模型反应,可能由于三唑和噻唑氮杂环卡宾的碱性不够,导致反应不能进行;
(2)咪唑氮杂环卡宾催化剂中4的效果最好,产率达到77%。
再以NHC4作为催化剂,考察质子溶剂(甲醇)、偶极溶剂(DMF)、非极性溶剂(二氯甲烷)等对反应的影响,结果表明:
(1)质子溶剂不利于反应的进行(entry10),由于溶液里质子浓度太高,不利于卡宾从硝基甲烷中夺取质子;
(2)可以从 entry8、entry9中可以看出,溶剂的极性对反应的影响很小,表明最好的溶剂是二氯甲烷,产率高达99%;
(3)我们对催化剂用量进行了优化,分别用17 mol%、10 mol%、10 mol%的4来催化反应,后两者的催化产率比17 mol%的低,由此得到最优的催化条件是以17 mol%的IPr4作为催化剂,二氯甲烷作为溶剂。
2.3 氮杂环卡宾催化Michael反应的底物拓展
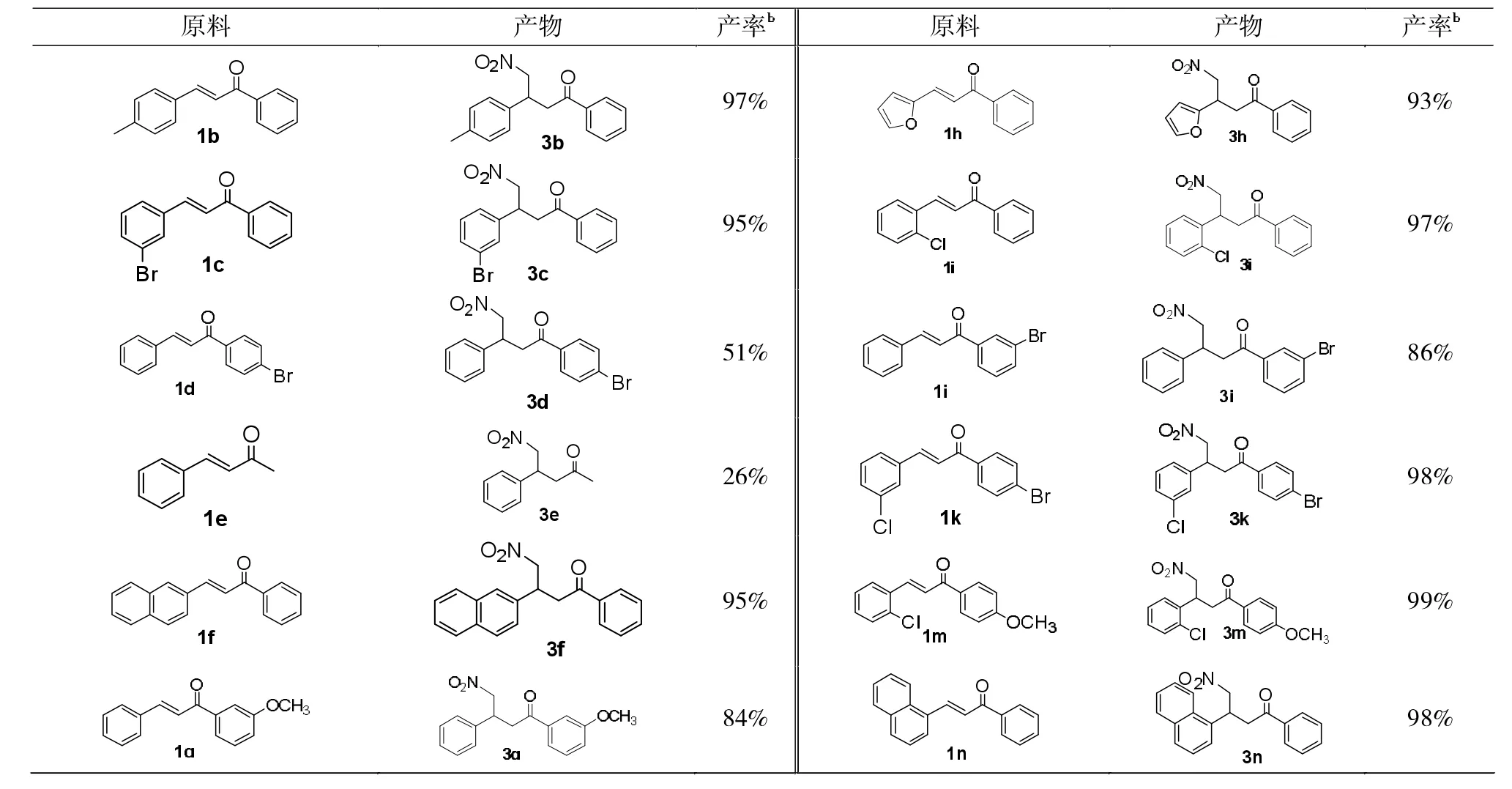
表2 氮杂环卡宾催化Micheal反应aTab.2 N-Heterocyclic carbene-catalyzed Micheal reactiona
在上述的最优的反应条件下,考察硝基甲烷与不同取代基查尔酮的Michael反应结果见表2。
由表2可以得出:
(1)芳香类的查尔酮可以很好适应该反应体系,产率高达99%,脂肪类的查尔酮也可以得到相应的Michael加成反应产物,但是产率不高,只得到26.6%,可能由于脂肪类查尔酮的双键不活泼,导致副产物增多,从而降低了反应的转化率。
(2)与羰基相连的苯环上的取代基,不论是供电子基还是拉电子基,都能得到相应的产物,并且得到84%-99%的产率。其原因可能是由于取代基的位置与双键离得比较远,导致对双键的影响降低;但是苯环上对位取代是溴时,只得到51.1%的产率,其原因可能是由于溴的拉电子能力强,导致副产物增多。不管双键上连的是吸电子基还是供电子基,都有利于反应的进行,产率为90%-99%,虽然苯环与双键共轭,但取代基对于双键的影响很小。另外,还考察了萘的查尔酮,也得到了很好的收率,这进一步说明苯环上的取代基对反应的影响不大。
结合理论和Scheidt、Huang以及我们实验室的工作,我们提出了一个可能的机理(如图1):首先,卡宾作为Brφnsted碱,具有夺质子能力,又因为硝基甲烷中硝基具有拉电子能力,所以甲基上的氢比较活泼,容易离去,所以硝基甲烷被NHC去质子形成中间体I,然后中间体对查尔酮的双键进攻,进行共轭加成产生中间体II,然后由于烯醇式的不稳定,异构化形成稳定的酮式结构,卡宾离去进行下一次循环,从而产生目标产物。其机理如图1所示:
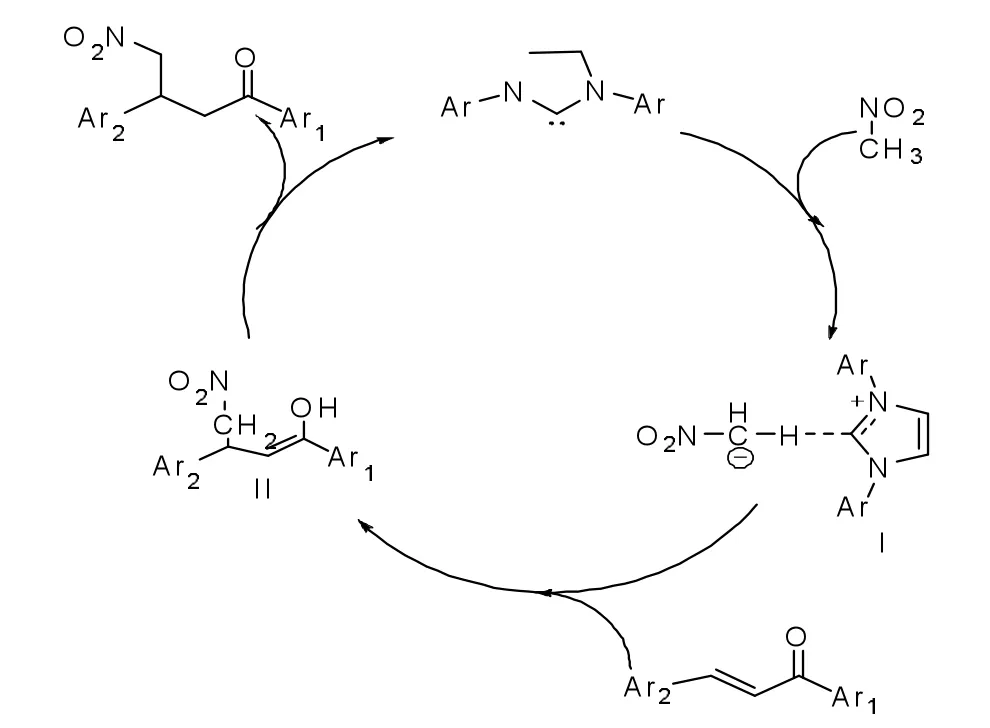
图1 反应催化机理示意图Fig.1 Reaction catalytic mechanism
3 结论
(1)本文利用环境友好催化剂,以及利用氮杂环卡宾的Brφnsted碱性质,成功催化了硝基甲烷对查尔酮的Michael加成反应,发展了一种新型的构筑C-C的新方法,同时,与以往催化Michael加成反应的催化剂相比,这种新方法使用的催化剂不含金属,且催化效率高。
(2)氮杂环卡宾催化Michael反应的最佳条件如下:以17 mol%(相对于查尔酮)4作为催化剂,0.3 mol硝基甲烷,0.2 mol的查尔酮,1 mL的二氯甲烷为溶剂,室温下反应12 h,以26%-99%的产率制备了14种硝基羰基化合物。
(3)不论查尔酮上是吸电子还是供电子基能适应反应体系,同样,不论苯环上是供电子基还是吸电子对反应的活性都有明显的提高,从而拓展了氮杂环卡宾催化的Michael加成反应。
[1]李院珍,王颖,贾晨辉,等.氮杂化卡宾催化的α-卤代烯醛的双硫化反应 [J].石河子大学学报 (自然科学版),2015,33(4):491-497.Li Y Z,Wang Y,Jia C H,L,et al.Doublevulcanization of N-heterocyclic carbine by aza hybrid carbine[J].Journal of Shihezi University(Natural Science),2015,33(4):491-497.
[2]He L,Guo H,Li Y Z et al.N-Heterocyclic Carbene-Catalyzed Formal Cross-Coupling Reaction of α-Haloenals with Thiols:Organocatalytic Construction of sp2 Carbon-Sulfur Bonds[J].Chem Commun,2014,50:3719-3724.
[3]Arduengo A J,Harlow R L,Kline M.A stable crystalline carbene[J].J Am Chem Soc,1991,113(1):361-364.
[4]Enders D,Kallfass U.An efficientnucleophilic carbene catalyst for the asymmetric benzoincondensation[J].Angew Chem Int Ed,2002,41,1743-1745.
[5]Kerr M S,Read de Alaniz,J.;Rovis,T.,A Highly Enantioselective CatalyticIntramolecular Stetter Reaction[J].J Am Chem Soc,2002,124(35),10298-10299.
[6]Zhang Y R,Lv,H,Zhou D,et al.Chiral N-Heterocyclic Carbene-Catalyzed Formal[4+2]Cycloaddition of Ketenes with Enones:Highly Enantioselective Synthesis of transandcis-δ-Lactones[J].Chem Eur J,2008,14,8473-8476.
[7]Li Y Z,Wang Y,Du G F,et al.N-Heterocyclic-Carbene-Catalyzed Sulfa-Michael additions[J].Asian J Org Chem,2015,4(4):327-332.
[8]Hao L,Du Y,Lv H,et al.Enantioselective activation of stable carboxylate esters as enolate equivalents via N-heterocyclic carbene catalysts[J].Org Lett,2012,14,2154-2157.
[9]He L,Lv H,Zhang Y R,et al,Formal cycloaddition of disubstituted ketenes with 2-oxoaldehydes catalyzed by chirral N-heterocyclic carbenes[J].J Org Chem,2008,73,8101-8103.
[10]Guin J,De Sarkar S,Grimme,S;Studer,A.BiomimeticCarbene-Catalyzed Oxidations of Aldehydes Using TEMPO[J].AngewChem,Int Ed,2008,47:8727-8730.
[11]Grasa G A,Kissling R M,Nolan S P.N-heterocyclic carbenes as versatile nucleophilic catalysts for transesterification/acylation reactions[J].Org Lett,2002,4:3583-3586.
[12]Boddaert T,Coquerel Y,Rodriguez J.Organocatalytic Activity of N-Heterocyclic Carbenes in the Michael Addition of 1,3-Dicarbonyl Compounds:Application to a Stereoselective SpirocyclizationSequence[J].Adv Synth Catal,2009,351:1744-748.
[13]Phillips E M,Riedrich M,Scheidt K A.N-Heterocyclic carbenecatalyzed conjugate additions of alcohols[J].J Am Chem Soc,2010,132:13179-13181.
[14]Chen J,Huang Y.Highly enantioselective sulfa-Michael add ition reactions using N-heterocyclic carbene as a noncovalent organocatalyst[J].Chem Sci,2015,6:4184-4189.
[15]Chen J,Huang Y.Asymmetric catalysis with N-heterocyclic carbenes as non-covalent chiral templates[J].Nat commun.2014,5:1-8.
[16]Wang Y,Du G F,Gu CZ,et al.N-heterocycliccarbene-catalysed vinylous-Mukaiyama-Michael reaction[J].Tetrahedron,2016,72(4):472-478.
[17]Wang Y,Du G F,Xing F,et al.N-Heterocyclic-Carbene-Catalysed Diastereoselective Vinylogous Mukaiyama/Michael Reaction of 2-(Trimethylsilyloxy)furan and Enones[J].Asian J Org Chem,2015,4(12):1362-1367.
[18]Gao H,He L.N-Heterocyclic Carbene-Catalyzed Direct Vinylogous Michael Reaction of β,γ-Unsaturated Butenolides[J].J Org Chem,2015,80:12606-12613.
[19]Wang Y,Xing F,Xue M,et al.N-Heterocyclic Carbene-Ca talyzed Vinylogous Mukaiyama Aldol Reaction of α-Keto Esters and α-Trifluoromethyl Ketones[J].Synthesis,2016,48(1):79-84.
[20]Guo H,Wang Y,Du G F,et al.N-Heterocyclic carbenecatalysed amidation of vinyl esters with aromatic amines[J].Tetrahedron,2015,71(21):3472-3479.
[21]郭豪,李院珍,王颖,等.乙烯酯与脂肪胺的酰胺化反应[J].石河子大学学报(自然科学版),2015,33(6):757-763.Gao H,Li Y Zh,Wang Y,et al.Ethylene amidation reaction with aliphatic amine[J].Journal of Shihezi University(Natural Science),2015,33(6):757-763.
[22]Liu P,Lei M,Ma L,et al.An Efficient Synthesis of 2-Aminofuran-3-carbonitriles via Cascade Stetter-γ-Ketonitrile Cyclization Reaction Catalyzed by N-Heterocyclic Carbene[J].Synlett,2011,42(37):1133-1136.
[23]Li Y,Xu H,Xing M,et al.Iodine-Promoted Construction of Polysubstituted 2,3-Dihydropyrroles from Chalcones and β-Enamine Ketones(Esters)[J].Org Lett,2015,17(15):3690-
N-heterocyclic carbene-catalyzed Michael reaction of nitro methane and chalcone
Xing Fen,Chen Lei,Feng Ze’nan1,Li Yangguo,Du Guangfen,He Lin*
(Key Laboratory for Green Processing of Chemical Engineering of Xinjiang Bingtuan/School of Chemistry and Chemical Engineering,Shihezi University,Shihezi,Xinjiang 832003 China)
As an important typical organic catalyst,N-heterocyclic carbenes (NHCs)has been widely used in organic synthesis in recent years.In this paper,we studied the Michael addition reaction of nitro methane and chalcone catalyzed by N-heterocyclic carbenes,which acted as a Brφnsted base in this process provides an effective method for the construction of γ-nitrocarbonylate.The results indicate that under the catalyst 17 mol%of IPr,in dichloromethane at room temperature,producing the desired products in 26%-99%yields.The mild conditions,environmental friendly,simple procedure,provide an effective method for synthesis γ-nitrocarbonylate.
N-heterocyclic carbenes;Michael reaction;Brφnsted base; γ-nitrocarbonylate;organic catalyst
O621.251;O643.36
A
10.13880/j.cnki.65-1174/n.2017.05.003
1007-7383(2017)05-0542-06
2017-03-14
国家自然科学基金项目(21662029)
邢芬(1990-),女,硕士研究生,专业方向为精细有机合成技术。
*通信作者:何林(1980-),男,教授,从事精细化学品合成,e-mail:helin@shzu.edu.cn。
