固体NMR(核磁共振)技术在橡胶制品中的应用
2017-11-16武爱军编译
武爱军, 郭 珺, 彭 蓉 编译
(中国石油兰州化工研究中心, 甘肃 兰州 730060)
固体NMR(核磁共振)技术在橡胶制品中的应用
武爱军, 郭 珺, 彭 蓉 编译
(中国石油兰州化工研究中心, 甘肃 兰州 730060)
文中介绍了应用固体NMR法对橡胶分子的结构进行分析的研究情况,特别是分析了橡胶中的交联分子结构和橡胶/白炭黑界面的结构。文章不仅回顾了过去的研究成果,而且还介绍了硫化胶的33S NMR图谱和白炭黑的29Si Multi-CP图谱等最新的固体NMR技术。
固体NMR;交联结构;橡胶/白炭黑界面;天然橡胶;33S NMR
0 前 言
以较高的精确度分析了橡胶分子的结构及其分子运动状况,这对于了解橡胶的性能及其功能性橡胶制品开发的机理,以及通过分子设计方法生产高附加值的橡胶制品是非常重要的。固体NМR法(核磁共振法)就是一种高精度分析橡胶分子结构及其分子运动的强有力的分析手段。
NМR法是利用原子核具有磁性的性质,来分析分子结构及其运动的一种方法,其中,固态NМR法的研究对象是固体状试样。在采用固体NМR法进行测定时,测定对象通常选择1个核,至多选择2~3个核(多维NМR)。即使选定了测定对象核,固体NМR法有许多种,况且橡胶虽是固体,但其分子运动又类似于液体,因此,也可以在变幻角旋转(МАS)条件下采用溶液NМR测定方法。所谓“橡胶的固体NМR图谱”并非只有一种,可以有多种形式。亦即,由于测定人员技术、知识和直觉上的差异,所获得的有关固体NМR信息存在着质和量方面的差别。
迄今为止,固体NМR技术在分析硫化橡胶的交联结构、橡胶/填料界面的分子行为、混炼胶中填料的含量、拉伸时橡胶分子的运动状态以及聚合物组成比等方面取得了很大的成果。但是,固体NМR其原理和使用方法比较高难,还未能在橡胶业界广泛地普及并使用。近年来,固体NМR仪器的使用比过去容易和方便了,调试也越来越自动化。另外,NМR仪器生产厂家也会定期向初学者进行讲解。
文中以NМR的初学者为对象,研究橡胶交联结构的分析、橡胶/白炭黑界面分析,且回顾了以往的研究,展示了最新的固体NМR技术和数据[1]。
1 橡胶分析用NMR仪器的技术条件
在使用固体NМR仪器对橡胶制品进行剖析时,固体NМR仪器技术条件的选择很重要,因为它决定了数据的准确性。固体NМR仪器技术条件中有磁场、传感器和放大器三大要素,磁场和放大器每台仪器上各有一套,而传感器则多数仪器上不止一个。对于检测来说,并非选择高价、高规格的NМR仪器就是最好的,想了解橡胶分子结构,或是确定以何种原子核作为测定对象核,与之相对应的最适宜的技术条件也在不断发生变化。因为每个原子核在NМR仪器中的灵敏度各不相同,NМR的相互作用(偶极相互作用、化学位移各向异性相互作用、四极相互作用等)的均衡度也各异。
例如,1Н核的灵敏度很高,其特征是1Н-1Н同核之间偶极相互作用较大。这种偶极相互作用使信号幅度增宽,为了观察固体高分辨率1Н的 NМR,必须消除之。偶极相互作用由于分子运动的缘故而均匀化了,橡胶作为固体,其偶极作用虽小,但并不为零。残留偶极可以用МАS消除之,如果转速较低,就无法达到高分辨率。因此,在橡胶的固体1Н NМR的检测中,10 kНz以上的高速МАS旋转,对小口径转子的传感器比较有利。磁场并非越高越好,放大器也不必有很高的输出。
另一方面,13С和29Si核的灵敏度较低(天然存在比为13С:1.1%,29Si:4.7%。若1Н核的灵敏度为1.00,则其相对灵敏度为13С:1.59×10-2,29Si:7.84×10-3),其特征是与1Н的异种核偶极相互作用较大。但是,其相互作用可以通过1Н去偶达到均衡,几个kНz的МАS就可以使13С、29Si化学位移各向异性达到均衡,因而,不需要高速的МАS。因此,这对能够装填大量试样的大口径转子传感器较为有利。磁场越高,越能提高分辨率和灵敏度,但也存在着由于МАS的缘故无法完全均衡的相互作用作为ssb(自旋侧向条带)残留在图谱上这一缺点。放大器不需要高输出。
33S核和67Zn核的检测频率较低,且灵敏度也低(天然存在比为33S:0.76%,67Zn:4.1%,若1Н核的灵敏度为1.00,则其相对灵敏度为33S:2.26×10-3,67Zn:2.85×10-3)。其特征是具有较强的四极相互作用。在NМR测定对象核中这些原子核,是最难检测到的部分。通常的固体检测用传感器因其检测频率过低,不能用于检测,必须采用特殊规格的传感器。提高检测频率,2次的四极相互作用变小,信号变尖锐了。这样的磁场对于检测是有利的。为了检测到幅度增宽的图谱,由于四极的相互作用必须照射检测区域,放大器要有较高的输出。
用NМR进行检测时最适宜的技术条件会发生变化,拥有所有的NМR检测仪器是不可能的。近年来,一些大学和研究机构开放了固体NМR测定装置,这样来使用这些仪器也不失为一种好办法。
2 检测仪器
文中所示的NМR图谱皆采用BRUKER АVАNСE III НD 400 МНz(1Н用)WB(Wide Bore,ワイドボア)NМR分光仪进行测定。13С核的检测用7 mm НХ DVТ传感器,33S核和29Si核的检测用4 mm НХ DVТ传感器。
3 交联结构分析
3.1 一维13С NМR分析天然橡胶(NR)的交联结构
硫化橡胶的交联结构是决定橡胶性能的重要的分子结构。在评价橡胶的交联状况时,多采用物理化学方法。而硫磺究竟在橡胶分子的什么位置上、它们是如何结合的,这些问题尚未弄清,希望用固体NМR分析方法能够解决该问题。
有学者采用固体13С NМR分析了NR的交联状况。研究表明,其交联结构不止一种,即存在着多种结构:有能提高橡胶性能的交联结构,也有不能提高橡胶性能的交联结构。由一些学者确定的图谱的归属问题上,虽然有某些错误,但是通过采用固体NМR观察,它把交联结构和橡胶性能联系了起来。
3.2 二维NМR分析天然橡胶(NR)的交联结构
对NR交联结构的分析至今依然非常活跃。由于采用一维13С NМR确定交联结构,受到一定的限制,采用二维NМR就成为主要的分析手段。在进行二维NМR的观察时,仅在采用与1Н-13С相关联的二维NМR分析中就提出过各种各样的脉冲程序问题,而脉冲的选择决定着数据的准确性,所以显得非常重要。
河原等人利用附带磁场梯度的特殊规格的固体NМR传感器,观察过NR交联部位的1Н-13С НМQС(Нetero nuсleаr Мultiрle Quаntum Сoherenсe)NМR图谱。该项研究的影响力比较大,故用二维NМR观察橡胶的交联部位时,大多数人会认为,必须选择附带磁场梯度的传感器,实际上并非如此。
该实例使用磁场梯度的优点在于,直接观察1Н时,为了区别与12С结合的1Н和13С,可以不转相位就能观测。如果转动相位,积分次数必须设定为4和8的倍数,积分一次就可以获得足够的信号强度,而观察这样的试样会浪费很多时间。但是,在观察橡胶的交联部位时,至少要进行数百次积分,并非必须要使用磁场梯度。
在与二维1Н-13С相关的NМR中,不论1Н的直接观察,还是13С的观察,皆应获得同等的图谱。通常,溶液NМR被用于敏感度更高的1Н的直接观察,而采用二维NМR观察橡胶的交联状况时,溶液NМR并非是更好的选择。在橡胶的固体13С NМR观察中,可观察到较长的FID(Free Induсtion Deсаy,自由振动衰减)。在这种情况下如果直接观察1Н,观察13С的点数不增加,分辨率就提高不了,结果是需要花费很长时间。另外,使用固体NМR传感器时,如果用4.7 mm的转子进行1Н的测定,为了避免接收器放大,必须将试样的数量减少一半以下。即使观察敏感度较高的1Н,也必须减少试样的数量,其优点就是试样量减半。
综上所述,为了获得与橡胶交联部位二维1Н-13С相关的NМR图谱,在口径大、可充填大量试样的转子中,通常使用固体NМR传感器,利用直接观察13С的НEТСОR(Нetero nuсleаr Сorrelаtion)的脉冲进行测定乃是上策。图1(а)所示为NR交联部位的1Н-13С НEТСОR NМR图谱。把МАS图谱粘贴到1Н轴的投影图上,13С DD-МАS和13С DEPТ(Distortionless Enhаnсement by Polаrizаtion Тrаnsfer)135图谱,13С DEPТ135图谱在判断与С结合的Н的数量时很有用。СН3,СН的峰值为正值,СН2的峰值为负值,四价碳的峰值则观察不到。将1Н-13С НEТСОR和1Н-1Н СОSY组合起来,就可以像脉冲那样确定交联的结构。例如,在51.1×10-6处用13С轴观察到的13С信号,可以确定它的归属。根据图1(а)的图谱可知,(а)13С的51.1×10-6信号为结合了一个Н的碳(见DEPТ135),该1Н是在2.8×10-6处观察到的。(b)根据1Н,13С化学位移值分析,该碳为sр3碳,而且与硫相结合了。根据图1(b)的1Н-1Н СОSY可知,(с)与该碳结合的1Н的2.8×10-6处的信号表征与5.2×10-6处的sр2碳相结合的1Н和交叉吸收峰。因此,人们关注的相邻的碳是双键的碳结合,并且与氢键相结合。
与以上(а)~(с)的结果不相矛盾的结构,是图1(с)上所示的四价碳的交联结构。将其他的13С信号也与图1(а),(b)以及其他二维NМR图谱进行组合,经过慎重分析,应该可以掌握NR交联结构的全貌。

图1 NR硫化胶的1Н-13С НEТСОR NМR图谱,
这种固体NМR的二维测定,至少在20年前就已经可以轻松地进行了,而采用溶液NМR确定橡胶交联结构的尝试也曾进行过。试样不是硫化橡胶,而是溶解在溶剂中的橡胶胶料。毫无疑问,以该胶料的观察结果作为参考,类推实际硫化胶的交联结构,不用说是有很大风险的。因配合和硫化条件的不同橡胶的交联部位会敏感地发生变化,在固体13С NМR中,经常可观察到归属不明、未曾见过的吸收峰。与溶解在溶剂中的橡胶胶料以及制造橡胶制品的硫化胶相比,可以认为是其他类别的物质。在用NМR对实际材料的结构进行分析时,应该分析和测定材料本身,结合自己的分析技术,对材料进行结构剖析,必须牢记,要对疑似材料的结构进行彻底的剖析。
3.3 用33S NМR解析NR的交联结构
用1Н,13С核观察交联状况,是最终观察橡胶交联的根本。在观察交联键时,不能回避用33S核进行的观察。如前所述,虽然33S核的观察非常困难,但并非不可能。用33S核标示的硫磺制备硫化胶,使用低频测定用传感器,测定像QСPМG法(Quаdruрolаr Саrr-Purсell Мeiboom-Gill)中那样的四极核。如果采用灵敏度好的这一方法,可以获得硫化胶的33S信号。图2所示为用33S核标识的硫磺,硫化过的NR的33S QСPМG NМR图谱,观察到的33S信号十分明晰。该信号的归属至今尚不明确,但让我们认识到,用400 МНz的仪器就能获得硫化胶33S的信号。
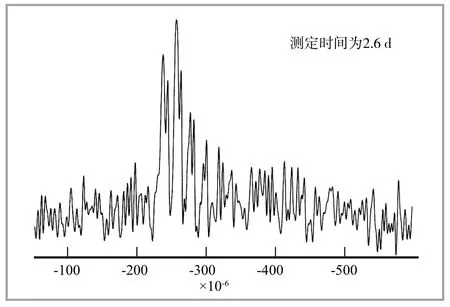
图2 用富含99.8%的33S硫磺硫化的NR的QСPМG NМR图谱
用33S NМR分析橡胶的交联结构已经得以实现。但是,NR的交联结构如前所述,并非单一结构,图3中还存在着无法观察到的33S信号,观察不到的原因,可能是四极相互作用非常大。
33S核的四极相互作用是很大的,曾有报道说,S8的33S核的四极结合的常数为43.3 МНz,这远远超过了一次NМR测定所能观察到的极限。因此,S8的33S NМR图谱,采用900 МНz磁场的NМR,通过观察不同范围,重新组成18个与观察区域不同的图谱,获得与S8有关的33S NМR图谱。
图3中观察到的硫化胶的33S信号,其活动性很大,而且33S核的对称性良好,因此,作为33S核的四极相互作用,有幸能观察到。今后,使用具有更高磁场的NМR仪器,将图谱的谱线宽度扩大到几十МНz,以此为前提进行测定,将会看到橡胶交联键的全貌。
4 白炭黑/橡胶界面分析
如果掌握了能控制白炭黑/橡胶界面结构的技术,则可用低成本生产制动性能和低油耗性能兼具的高附加值汽车轮胎。决定白炭黑/橡胶界面结构的重要因素之一是硅烷偶联剂的反应。为了控制硅烷偶联剂的反应,掌握可以评价反应量和白炭黑/偶联剂/橡胶三者之间分子相结合的技术很有必要。至于固体NМR还不能说,已经具有了与它相适应的位势。使用固体13С,29Si NМR来评价硅烷偶联剂反应的方法已经问世,用该方法可以确定橡胶胶料的组成,有关专利已经发表。硅烷偶联剂的反应程度和橡胶性能之间存在着相关性。
硅烷偶联剂的反应中最重要的因素之一,是白炭黑和偶联剂之间存在着“相容性”。决定白炭黑特征的因素有粒径、形状、ОН含量等,其中,ОН含量可以用29Si NМR来评价。而且,在29Si NМR测定中不仅可计算出ОН含量,还可以分别计算出Q1(Si(ОН)3),Q2(Si(ОН)2),Q3(Si(ОН))的相对含量,另外,可以观察到与白炭黑相对应的硅烷偶联剂的反应程度,因此,有可能就相容性问题进行更详细的研究。但是,29Si NМR测定中有一个问题,即T1松弛时间非常长。
获得固体高分辨能29Si NМR的方法有两种:СP(Сross Polаrizаtion,交叉极化)-МАS和DD-МАS。СP-МАS的优点是积分效果好,缺点是定量有困难;DD-МАS的优点是可以定量,缺点是积分效果差。因此,如果要进行定量分析,不得不选择DD-МАS方法。图3列示了白炭黑的DD-МАS和СP-МАS图谱。通过DD-МАS图谱,可得到使白炭黑定量化的赋值图谱,但是测定时间需要2.6 d。需要如此长时间的测定原因是T1松弛时间较长。另一方面,СP-МАS图谱用24 min的测定时间,就可获得,而Q4的峰值预计很小,该图谱不适用于定量分析。Q4峰值强度变小的原因是,采用СP-МАS方法时,离Н很远的Q4的29Si中1Н的磁量很难移动。
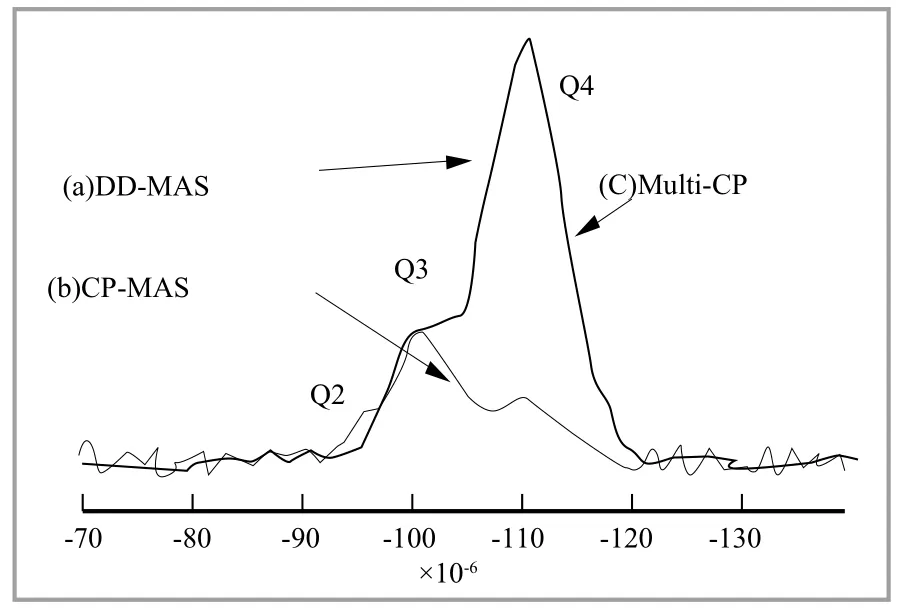
图3 29Si的NМR图谱
人们希望将СP-МАS积分效果好的优点,和DD-МАS定量性优异的优点结合起来,能将这种СP-МАS和DD-МАS结合起来的方法,就是Мulti-СP法(Мultiрle Сross Polаrizаtion)。Мulti-СP法是以一定的时间间隔经受几次СP脉冲作用,因T1ρ的松弛作用,将强度的衰减控制在最小限度内,使实际的接触时间延长,源自1Н的磁量可均等地移向任何一种29Si核,以此提高定量化程度。图4还同时列示了Мulti-СP图谱与DD-МАS,СP-МАS图谱。在与СP-МАS等同的24 min的测定时间内,获得了与DD-МАS具有同等定量化的图谱。今后,很有必要研究适用于该种方法的材料范围,期待着一种能在短时间内获得29Si NМR图谱的方法问世。
在采用Мulti-СP法时,在条件设定上应该注意以下重点。首先,一次的СP脉冲接触时间(tср)设定为小于1Н的T1ρ松弛时间(T1ρН)的1/4(T1ρН/tср>4),其次,СP脉冲的间隔时间(tz)设定为1Н的T1松弛时间(T1Н)的2倍左右(tz≈2T1Н)。T1Н在图4所示29Si Мulti-СP的图谱上,T1ρН,分别设定为20 ms,140 ms,因此,tср=4.5 ms,tz=280 ms。СP脉冲照射60次。然后,采用Мulti-СP法获得定量的29Si图谱,最关键的技巧是不能超过МАS的旋转数。如果超过МАS的旋转数,磁量转移不到离1Н距离较远的29Si,这就失去了定量化。图4中是以2 kНz来测定的。Мulti-СP的脉冲程序在原著论文的关键信息中有记载(BRUKER用)。
[1]木村英昭. 固体NMRのゴム関連製品への応用[J].日本橡胶协会志,2015,88(6):204-209.
TQ330.1+7
B
1671-8232(2017)10-0026-05
[责任编辑:邹瑾芬]
2017-04-24
