通过有机配体中四嗪基团的原位水解提高金属有机框架的CO2吸附性能
2017-11-13钱彬彬卜显和
钱彬彬 赵 萌 常 泽*, 卜显和*,,2
通过有机配体中四嗪基团的原位水解提高金属有机框架的CO2吸附性能
钱彬彬1赵 萌1常 泽*,1卜显和*,1,2
(1南开大学材料科学与工程学院,国家新材料研究院,天津市金属与分子基材料化学重点实验室,天津 300350)
(2南开大学化学学院,先进能源材料化学教育部重点实验室,天津 300071)
在保持原有“层-柱”MOF,[Zn4(bpta)2(dipytz)2(H2O)2]·4DMF·H2O (1)(H4bpta=2,2′,6,6′-联苯四羧酸,dipytz=3,6-二(4-吡啶基)-1,2,4,5-四嗪)主体结构不变的情况下,通过dipytz配体中四嗪环的原位水解反应将极性的二芳酰基联氨基团引入框架,成功构筑出配合物[Zn4(bpta)2(dipytzhydr)2(H2O)2]·solvent(2)(dipytzhydr=1,2-二异烟酰基肼)。 对配合物 2 的系统表征和气体吸附性质研究结果证实了功能化目标的实现:配合物2相比于配合物1展现出更高的二氧化碳吸附热(由28.8 kJ·mol-1升高至30.3 kJ·mol-1)和CO2/CH4吸附选择性。以上结果表明基于配体中四嗪基团的原位水解后修饰能够有效提高相关MOFs材料的CO2吸附性能。
金属有机框架;四嗪;后合成;性能调控;CO2吸附
0 Introduction
With the rapid development of modern industry,CO2release from burning fossil fuels has been a global environmental issue[1]and it calls for much efforts on the capture and separation of CO2[2-4].Among the various strategiesforCO2capture,physical adsorption with porous materials is considered to be one of the effective methods[5],and the development of porous materials with high CO2adsorption capacity has attracted much attention in recent years[6-7].
Due to the adjustable pore size,high porosity and large surface area,metal-organic frameworks (MOFs)has become a promising candidate for gas adsorption and separation[8-15].The strategies for improving CO2adsorption capacity and selectivity of MOFs include the incorporation of unsaturated metal cation centers,metal doping and chemical functionalization.Among them,chemical functionalization has been proved to be simple and efficient due to the diversity of functionalized groups[16-17].
Among numerous organic ligands,tetrazine derivative ligands are widely used in function-oriented construction of MOFs[18-24],and the tetrazine moiety can be hydrolyzed to form polar acyl hydrazine groups[25-26].This feature can help to enhance the affinity toward CO2and promote the adsorption and separation performance of the corresponding MOFs.However,in contrast to the rigid aromatic moiety of the parental tetrazine moiety,the flexible backbone of the acyl hydrazine moiety may affect the assembly of porous MOFs with desirable structure,which could limit the application of direct synthesis method.On the other hand,as an alternative method,the post-synthesis insitu hydrolysis of tetrazine moiety into acyl hydrazine requires high stability and strong resistance toward moisture,which is difficult to meet for most MOFs.Therefore,it is our interest to overcome the disadvantage and achieve the targeted introduction of acyl hydrazine moiety into MOFs from hydrolysis of tetrazine for enhanced CO2sorption and separation.
Based on our previous studies,the “pillar-layer”strategy,where the “layers”are composed of 1,1′-biphenyl-2,2′,6,6′-tetracarboxylic acid (H4bpta)and Zn2+ions and the bipyridine ligands serve as “pillar”,has been proved to be effective for the targeted construction of porous framework with distinct sorption behaviors[27-28]. In this system,the utilization of pillar ligand di-3,6-(4-pyridyl)-1,2,4,5-tetrazine (dipytz)can result in desired “pillar-layer” structure [Zn4(bpta)2(dipytz)2(H2O)2]·4DMF·H2O and can tune the pore geometry[28],as is estimated from the powder X-ray diffraction patterns.Considering the feature of tetrazine moieties,[Zn4(bpta)2(dipytz)2(H2O)2]was chosen and the post-synthesis in-situ hydrolysis modification is achieved.Herein,we report the structure of [Zn4(bpta)2(dipytz)2(H2O)2]·4DMF·H2O (1)and the construction of[Zn4(bpta)2(dipytzhydr)2(H2O)2]·solvent (2)(dipytzhydr=1,2-diisonicotinoylhydrazine)from complex 1 through post-synthesis in-situ hydrolysis modification(Scheme 1).Due to the moisture stability of the framework of complex 1,the porous “pillar-layer” framework is well retained afterthe modification process,and the resulted complex 2 reveals enhanced affinity toward CO2compared to that of complex 1.The presence of polar acyl hydrazine moiety in complex 2 results in better CO2/CH4selectivity as expected.
1 Experimental
1.1 Materials and methods
All solvents and chemicals for synthesis were obtained commercially and used withoutfurther purification.H4bpta and dipytz were synthesized according to the reported methods[29-30].The powder X-ray diffraction (PXRD)patterns were recorded by a Rigaku Miniflex 600 diffractometer at 40 kV and 15 mA with a Cu Kα radiation (λ=0.154 18 nm)and a graphite monochromator in the range of 3°~50°.Thermogravimetric analysis (TGA)was performed with a Rigaku standard TG-DTA analyzer with heating rate of 10℃·min-1between room temperature and 800℃ in air;empty Al2O3crucible was used as reference.IR spectra were carried out on a Tensor 37 (Bruker,German)FT-IR spectrometer in the range of 400~4 000 cm-1using KBr pellets.Elemental Analysis (C,H and N)were performed on a Vario EL cube analyzer.

Scheme 1 Structure of ligands,synthesis process of complexes and the corresponding images of the products
1.2 Synthesis of the complexes
[Zn4(bpta)2(dipytz)2(H2O)2]·4DMF·H2O (1).Zn(NO3)2·6H2O (0.1 mmol),H4bpta (0.05 mmol)and dipytz(0.05 mmol)were added to a DMF/ethanol mixture (10 mL,VDMF/VEtOH=1),sealed in a capped vial and ultrasonicated for 30 min.Then the vial was kept at 80℃for 24 h.Pale red bulk crystals were collected by filtration,then washed with DMF,and dried in air(Yield:70%based on H4bpta).FT-IR (KBr pellets,cm-1):3 855 w,3 742 m,3 423 s,2 362 m,2 322 m,1 835 w,1 601 s,1 550 s,1 459 m,1 386 s,1 220 w,1 159 w,1 063 w,1 025 w,924 w,839 m,777 m,715 m,601 m,534 w,454 w.Anal.Calcd.for C68H62N16O23Zn4(%):C,47.13;H,3.61;N,12.93.Found(%):C,47.31;H,3.30;N,12.90.
[Zn4(bpta)2(dipytzhydr)2(H2O)2]·solvent (2).Complex 2 was produced by heating complex 1 in water at 80℃for 12 h.Yellow crystalline powder were collected by filtration,washed with H2O and dried in air (Yield:98%,based on complex 1).FT-IR (KBr pellets,cm-1):3 925 w,3 897 w,3 862 w,3 743 w,3 423 s,2 362 m,2 333 m,1 917 w,1 868 w,1 837 w,1 677 m,1 611 s,1 551s,1 458 m,1 372 s,1 294 m,1 222 w,1 154 w,1 102 w,1 067 w,1 028 w,930 w,835 m,778 m,713 m,666 m,589 w,539 w,451 w,422 w.
1.3 Crystal structure determination
Single-crystal X-ray diffraction measurement was conducted at BL16B1 beamline at Shanghai Synchrotron Radiation Facility (SSRF)at 113 K.The determinations of unit cell parameters and data collections were performed with Mo Kα radiation (λ=0.071 073 nm),and unit cell dimensions were obtained with least-squares refinements.The structure of 1 was solved by direct methods using the SHELXS of the SHELXTL and refined by SHELXL[31].Zinc atoms in 1 were located from the E maps,and other nonhydrogen atoms were located in successive difference Fourier synthesis and refined anisotropically.The hydrogen atoms were added theoretically,riding on the concerned atoms,and refined with fixed thermal factor.The final refinement was performed by fullmatrix least-squares methods with anisotropic thermal parameters for non-hydrogen atoms on F2.The solvent molecules in 1 were disordered and could not be modeled properly,so the contribution of disordered solvent molecules were removed by SQUEEZE in PLATON[32]and the results were appended in the CIF file.Detailed crystallographic data were summarized in Table S1,and the selected bond lengths and angles are given in Table S2 and S3(Supporting Information).CCDC:1567303,1.
1.4 Gas sorption measurements
Gas sorption measurements were performed with an ASAP 2020 M gas adsorption analyzer.UHP-grade gases were used in measurements.The N2sorption isotherm measurements were proceeded at 77 K.The CO2and CH4sorption isotherm measurements were carried out at 273 and 298 K,respectively.
Before measurements,the samples were soaked in anhydrous methanol for 3 days to exchange solvent molecules in the channels and then filtrated and dried at room temperature.Activation of the methanolexchanged samples was performed under high vacuum(less than 1.33 mPa)at 50 ℃ overnight.About 100 mg of the desolvated samples were used for gas sorption measurements.
2 Results and discussion
2.1 Synthesis of complexes
Complex 1 was synthesized based on the reported method[28],while single-crystal suitable for X-ray diffraction analysiswereobtained.Therefore,the structure of complex 1 was determined straightforwardly.
To achieve the post-synthesis in-situ hydrolysis modification of complex 1,various methods have been tried,and it was found that direct heating of complex 1 in water could be a straightforward way,proved by the fading of the characteristic red color of tetrazine moiety of the sample originated from the opening of rings.It should be noted that the crystallinity of the sample is well retained after the hydrolysis.This should be attributed to the relatively high moisture stability of the Zn4(bpta)2(H2O)2layer structure and the Zn-N coor-dination bonds that could survive the hydrolysis reaction conditions.As a result,complex 2 was obtained as expected.Though no suitable crystal for single-crystal X-ray diffraction analysiswasobtained forcomplex 2,the high crystallinity just benefits its further characterization and properties investigations.
2.2 Structure determination
Single-crystal X-ray diffraction analysis reveals that complex 1 crystallizes in the monoclinic space group C2/c.As shown in Fig.1a,there are two types of crystallographically independent Zn2+ions,one bpta4-ligand,one dipytz ligand and one H2O molecule in the asymmetricunitof1.The Zn1 centerisfourcoordinated by three carboxylate oxygen atoms from three different bpta4-ligands and one nitrogen atom from one dipytz ligand to form a tetrahedral geometry.The Zn2 center adopts a six-coordinated distorted octahedral geometry,completed by four O atoms of three carboxylate groups from two bpta4-ligands,one O atom from the terminal H2O,and one N atom from one dipytz ligand.In the direction of the a axis,twodimensional (2D)layers are constructed by bpta4-ligands connecting with Zn2+ions.Each bpta4-coordinates to five Zn2+centers through four carboxylate groups by mondentate or bidentate coordination modes(Fig.1b).Furthermore,dipytz ligands serve as “pillar”and connect adjacent layers to form three-dimensional(3D)“pillar-layered” frameworks,which contain channels alone the b and c axis (Fig.1c and 1d).The channels are filled with solvent molecules.PLATON analysis showed that the accessible volume of 1 is 38.9%of the crystal volume(3.138 9 nm3out of 8.076 0 nm3for unit cell volumes)after removal solvent molecules in frameworks.It should be noted that the cell parameters determined herein consist well with that obtained from PXRD patterns[28],and the increased porosity of complex 1 compared with other complexes based on shorter pillar ligands suggests that the application of longer ligand could benefit the formation of porous framework.

Fig.1 Crystal structure of 1:(a)Coordination environment of Zn2+ions;(b)2D layer assembled by Zn2+ions and bpta4-ligands viewing along the a direction;(c)Channels along b direction;(d)Channels along c direction
As no suitable crystal of complex 2 for singlecrystal X-ray diffraction measurement was obtained,the structure aswellasthe componentofthe crystalline product was determined by comprehensive characterization by FT-IR and PXRD.As mentioned in the synthesis part,the complete fading of the red color of complex 1 in the hydrolysis reaction can be ascribed to the ring opening of tetrazine moiety in the dipytz ligand and the corresponding formation of acyl hydrazine group.Beside the change of color,this reaction can also be proved by the FT-IR spectra:two new characteristic absorption peaks appear in the location of 1 677 and 1 294 cm-1in the spectrum of complex 2 compared with that of complex 1 (Fig.2),which should be attributed to the presence of carbonyl and imino groups in the sample from the hydrolysis of tetrazine.Furthermore,the elementalanalysisof complex 2 after removal of solvent at 150℃also confirms the result.The experimental values (C,47.47;H,2.93;N,7.58)matches the theoretical values(calculated as C56H35N8Zn4O21.5,C,47.18;H,2.47;N,7.86)well,which directly proves the occurrence of the hydrolysis reaction.All these results suggest the complete hydrolysis of dipytz and the formation of dipytzhydrduring the post-synthesis reaction.On the other hand,the similar PXRD pattern indicates that complex 2 possessesthe similar “pillar-layered”structure of 1 (Fig.3)with the dipytzhydrligand serve as pillars.Then the framework of complex 2 could be assigned as Zn4(bpta)2(dipytzhydr)2(H2O)2according to the component and structure of complex 1.
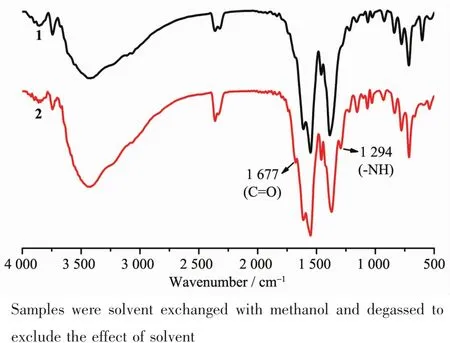
Fig.2 FI-IR spectra of complexes 1 and 2
2.3 Phase purity and stability investigations
PXRD was performed to confirm the phase purity and stability of complexes 1 and 2 after solvent exchange and degas process,as shown in Fig.3.For 1,the pattern of as-synthesized sample matches well with the simulated pattern from single-crystal data,suggesting the phase purity of the bulk sample.While forcomplex 2,the PXRD patternsofthe assynthesized sample is nearly the same as that of complex 1,indicating the structural similarity of these two complexes.Furthermore,solvent-exchanged and activated samples of both complexes are still crystalline,indicating the good stability of complexes.The shift of peaks and appearance of new peaks should be attributed to the distortion of the crystal lattice in response to removal of guest molecules,which iscommonly observed in many MOFs[28].Interestingly,the peak displacement of complex 2 after degassed at 50℃is more significant than that of complex 1.This phenomenon should be attributed to the more flexible backbone of dipytzhydrcompared with thatofdipytz.The hydrolysisreaction ofrigid tetrazine ring result in acyl hydrazine group with nonrigid backbone.Accordingly,the framework of complex 2 should be more flexible and more responsive to the removal of guest molecules.It should be noted that when the samples of complexes 1 and 2 were exposed to air after gas adsorption experiments,the patterns could restore well.These results suggest the structure transformation is reversible upon the removaland recoveryofguestmolecules,which should be originated from the flexibility of the pillar ligands and the framework.
TGA experiments reflect the thermal stability of materials (Fig.S1 and S2,Supporting Information).The TG curve of complex 1 shows weight loss of 12.1%from 30 to 140℃corresponding to the loss of solvent molecules.A sustained weight loss followed from 140 to 210℃responses to hydrolysis of dipytz with ring opening.However,after the sample was soaked in methanol for 3 days,hydrolysis is prevented.What′s more,the loss of coordinated H2O occurs at 163 ℃and the framework could be stable up to 340℃.
The TG curve of complex 2 shows 20%weight loss before 100℃,which should be assigned to the removal of solvent molecules.Then it reveals a weight loss of 3%in the temperature range of 100~177 ℃,which responses to the weight of coordinated H2O molecules.Also,the activated sample 2 shows high thermal stability up to 340℃,indicating the high thermal stability of these “pillar-layered” MOFs.
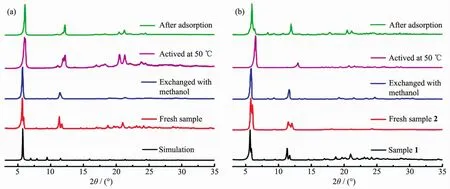
Fig.3 PXRD patterns of complexes 1 (a)and 2 (b)
2.4 Adsorption studies
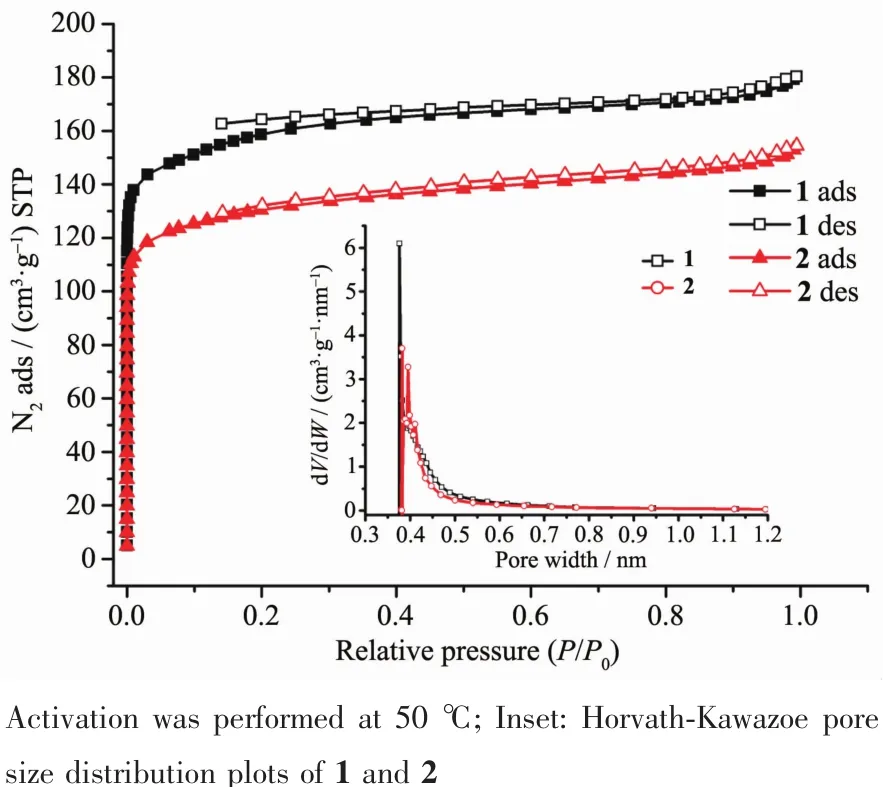
Fig.4 Isotherms of N2at 77 K for 1 and 2
Nitrogen gas adsorption experiments at 77 K were performed to investigate the porosity of complexes 1 and 2 (Fig.4).All samples were activated at 50℃ before the measurements.As shown in Fig.4,both the N2sorption isothermsofthe activated samples of 1 and 2 illustrate fully reversible typeⅠisotherms,which indicates the microporous structure of the complexes.The saturation uptake is 180.6 cm3·g-1for 1 and 155.2 cm3·g-1for 2,respectively.The total microporous volume is calculated to be 0.28 cm3·g-1for 1 and 0.24 cm3·g-1for 2 (calculated by Horvath-Kawazoe model), while the pore size distributions are almost identical (Fig.4 inset).These sorption isotherms were analyzed by the methods of Brunauer-Emmett-Teller (BET)and Langmuir.The apparent BET and Langmuir surface areas are 514 and 712 m2·g-1for 1 and 420 and 586 m2·g-1for 2,respectively.Considering the conclusions from PXRD discussions,the reduced surface area and pore volume of complex 2 compared to those of 1 should be attributed to the shrink of flexible framework in response to the removal of guest molecules.
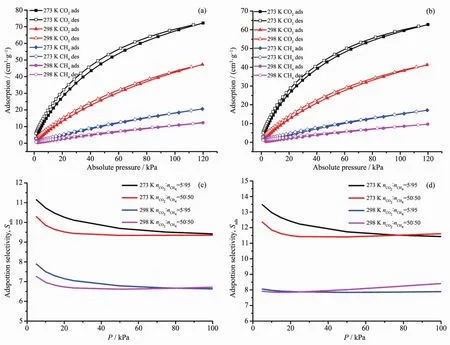
Fig.5 CO2and CH4isotherms of 1 (a)and 2 (b)at 273 and 298 K;IAST adsorption selectivity of 1 (c)and 2 (d)for CO2/CH4at 273 and 298 K
On the basis of previous research,the presence ofcarboxamidegroup in MOFsmaybenefitits selectivity adsorption performance toward CO2due to the enhanced gas-framework affinity.Therefore,CO2adsorption isotherms of 1 and 2 were recorded at 273 and 298 K,respectively,to evaluate the effect of postsynthesis modification on CO2sorption and separation(Fig.5a and 5b).The maximum CO2uptake of 1 reaches to 72.2 cm3·g-1at 273 K,120 kPa and 47.3 cm3·g-1at 298 K,120 kPa.For 2,the CO2maximum adsorption amounts are 62.8 cm3·g-1at 273 K ,120 kPa and 41.3 cm3·g-1at 298 K,120 kPa.The differences between the CO2uptakes of complexes 1 and 2 under the same conditions fit their differences in pore volume determined from N2sorption well.To further evaluate the interaction between the adsorbed CO2molecules and the frameworks,the CO2adsorption enthalpies (Qst)of 1 and 2 are calculated using the Virial equation by fitting adsorption isotherms at 273 and 298 K (Fig.6).The CO2Qstvalue of 1 is 28.8 kJ·mol-1at zero loading,while the initial Qstvalue of 2 increasesto 30.3 kJ·mol-1,which indicatesa relatively stronger interaction between CO2and the framework of complex 2.It should be noted that Qstvalue of complex 2 surpass that of complex 1 in the whole range of loading.This should be attributed to the readily accessible polar acyl hydrazine sites on the pore surface defined by the backbone of dipytzhydrpillar,which could be accessed in the whole gas sorption procedure due to the limited pore dimension.These results prove that the post-synthesis in-situ hydrolysis of tetrazine could be an effective method for the targeted modification of MOFs toward CO2sorption.

Fig.6 CO2adsorption enthalpies (Qst)of complexes 1 and 2
In addition to the evaluation of Qst,the ideal adsorbed solution theory (IAST)calculation was also employed to further evaluate the effect of modification on selective CO2adsorption over CH4.The binary CO2/CH4mixtures with molar ratio of 5∶95 and 50∶50 were selected as model system.To establish the relationship between the CH4uptake and pressure for calculation,the CH4adsorption isotherm were also measured at 273 and 298 K.The maximum CH4uptakes of 1 are 20.6 cm3·g-1at 273 K and 12.3 cm3·g-1at 298 K.For 2,the values are 17.1 cm3·g-1at 273 K and 9.7 cm3·g-1at 298 K,respectively.Then,the single-component isotherms of CO2and CH4at 273 and 298 K were fitted with Langmuir-Freundlich equation (Fig.S3,Supporting Information),and the fitting parameters(Table S4)are used for the IAST calculations.
As shown in Fig.5c,the calculated selectivity of CO2over CH4for complex 1 are 11.1 (nCO2∶nCH4=5∶95),10.3 (nCO2∶nCH4=50∶50)at 273 K and 7.9 (5∶95),7.3 (50∶50)at 298 K.While the calculated CO2/CH4selectivity of complex 2 are 13.5 (5∶95),12.4 (50∶50)at 273 K and 8.1 (5∶95),7.9 (50∶50)at 298 K as shown in Fig.5d.Obviously,the CO2/CH4selectivity of complex 2 are enhanced compared with that of complex 1,which in line with the results calculated from Qst.For complexes 1 and 2,the relatively higher selectivity occurs at lower pressure and lower temperature,which benefiting from the improved CO2storage capacities and enhanced CO2binding affinity of frameworks.
3 Conclusions
In summary,the post-synthesis in-situ hydrolysis of tetrazine moiety in MOFs was investigated for improving CO2sorption performance.With this method,[Zn4(bpta)2(dipytz)2(H2O)2]·4DMF·H2O (1)was successfully modified into [Zn4(bpta)2(dipytzhydr)2(H2O)2]·solvent(2)with polar acyl hydrazine groups,of which the “pillar-layer” framework structure is well retained.Complex 2 represents enhanced CO2-framework affinity and CO2/CH4selectivity compared with that of complex 1,as expected.The achievement in this work provides a valuable strategy for the targeted construction and modification of MOFs toward CO2sorption applications.
Acknowledgments:This work was supported by the National Natural Science Foundation of China (Grants No.21531005,21421001,21671112)and Natural Science Fund of Tianjin,China (Grant No.15JCZDJC38800).We thank the staffs from BL16B1 beamline of National Facility for Protein Science in Shanghai(NFPS)at Shanghai Synchrotron Radiation Facility,for assistance during data collection.
Supporting information is available at http://www.wjhxxb.cn
[1]D′Alessandro D M,Smit B,Long J R.Angew.Chem.Int.Ed.,2010,49:6058-6082
[2]Jacobson M Z.Energy Environ.Sci.,2009,2:148-155
[3]Sumida K,Rogow D L,Mason J A,et al.Chem.Rev.,2012,112:724-781
[4]Nugent P,Belmabkhout Y,Burd S D,et al.Nature,2013,495:80-84
[5]Haszeldine R S.Science,2009,325:1647-1652
[6]Christopher W J,Edward J M.ChemSusChem,2010,3:863-864
[7]Rubin E S,John H,Marks A,et al.Prog.Energy Combust.Sci.,2012,38:630-671
[8]Wang H,Xu J,Bu X H,et al.Angew.Chem.Int.Ed.,2015,54:5966-5699
[9]Tian D,Chen Q,Bu X H,et al.Angew.Chem.Int.Ed.,2014,53:837-849
[10]Chang,Z,Yang D H,Bu X H,et al.Adv.Mater.,2015,27:5432-5435
[11]Chen K J,Chen X M,Zaworotko M J,et al.Angew.Chem.Int.Ed.,2016,55:10268-10272
[12]Lin R B,Li T Y,Chen X M,et al.Chem.Sci.,2015,6:2516-2521
[13]Wang L,He C T,Chen X M,et al.J.Am.Chem.Soc.,2017,139:8086-8089
[14]Chen C,Wei Z,Su C Y,et al.Angew.Chem.Int.Ed.,2016,55:9932-9937
[15]Liu D,Chang Y J,Lang J P.CrystEngCommun,2011,13:1851-1857
[16]Liu B,Yao S,Liu Y L,et al.Chem.Commun.,2016,52:3223-3229
[17]Luo X L,Cao Y,Liu Y L,et al.J.Am.Chem.Soc.,2016,138:2969-2972
[18]Yao S,Wang D M,Liu Y L,et al.J.Am.Chem.Soc.,2015,3:16627-16631
[19]Philipp M F,Wisser V B,Stefan K,et al.Chem.Mater.,2015,27:2460-2467
[20]Li C,Ge H,Li J L,et al.RSC Adv.,2015,5:12277-12286
[21]Zhang Z X,Ding N N,Zhang W H,et al.Angew.Chem.Int.Ed.,2014,53:4628-4632
[22]John E C,Jason R P,Cameron J K,et al.Angew.Chem.Int.Ed.,2014,53:10164-10168
[23]Lu Z Z,Zhang R,Zheng H G,et al.J.Am.Chem.Soc.,2011,133:4172-4174
[24]Yoshihiro Y,Yuya H,Takahiro I,et al.J.Am.Chem.Soc.,2010,132:9555-9557
[25]Vagin S I,Ott A K,Rieger B,et al.Chem.Eur.J.,2009,15:5845-5853
[26]Bu X H,Liu H,Shionoya J,et al.Inorg.Chem.,2002,41:1855-1861
[27]Xuan Z H,Chang Z,Bu X H,et al.Inorg.Chem.,2014,53:8985-8990
[28]Chang Z,Zhang D S,Bu X H,et al.Inorg.Chem.,2011,50:7555-7562
[29]Peter H D,Mary E W,Charlotte L S,et al.J.Am.Chem.Soc.,2010,126:12989-13001
[30]Suh M P,Moon H R,Lee E Y,et al.J.Am.Chem.Soc.,2006,14:4710-4718
[31]Sheldrick G M.Acta Crystallogr.Sect.A:Found.Crystallogr.,2008,A64:112-122
[32]Spek A L.J.Appl.Crystallogr.,2003,36:7-13
Enhanced CO2Sorption Performance of Metal-Organic Frameworks by in-Situ Hydrolysis of Tetrazine Moiety in the Ligand
QIAN Bin-Bin1ZHAO Meng1CHANG Ze*,1BU Xian-He*,1,2
(1School of Materials Science and Engineering,National Institute for Advanced Materials,TKL of Metal and Molecule-Based Material Chemistry,Nankai University,Tianjin 300350,China)
(2College of Chemistry,Key Laboratory of Advanced Energy Materials Chemistry
(Ministry of Education),Nankai University,Tianjin 300071,China)
The polar acyl hydrazine groups were introduced into a “pillar-layer” MOF,[Zn4(bpta)2(dipytz)2(H2O)2]·4DMF·H2O (1)(H4bpta=1,1′-biphenyl-2,2′,6,6′-tetracarboxylic acid and dipytz=di-3,6-(4-pyridyl)-1,2,4,5-tetrazine),through the in-situ hydrolysis modification of the dipytz pillar ligand,and [Zn4(bpta)2(dipytzhydr)2(H2O)2]·solvent (2)(dipytzhydr=1,2-diisonicotinoylhydrazine)was obtained.The results of gas sorption measurement show that complex 2 has an enhanced CO2-framework affinity (initial CO2adsorption enthalpies increase from 28.8 to 30.3 kJ·mol-1)and higher CO2/CH4selectivity compared with that of complex 1.The present work indicates that in-situ hydrolysis modification is highly potential for the enhancement of CO2adsorption performance of tetrazine functionalized MOFs.CCDC:1567303,1.
metal-organic frameworks;tetrazine;post-synthesis;property modulation;CO2sorption
O614.24+1
A
1001-4861(2017)11-2051-09
10.11862/CJIC.2017.251
2017-08-22。收修改稿日期:2017-09-29。
国家自然科学基金(No.21531005,21421001,21671112)和天津市自然科学基金(No.15JCZDJC38800)资助项目。
*通信联系人。E-mail:changze@nankai.edu.cn,buxh@nankai.edu.cn
