基于RAD-seq技术的异型花SSR信息分析
2017-11-10王久利朱明星徐明行陈世龙张发起
王久利 朱明星 徐明行 陈世龙 张发起*
(1.中国科学院高原生物适应与进化重点实验室,中国科学院西北高原生物研究所,西北生态环境资源研究院,西宁 810001; 2.中国科学院大学,北京 100039; 3.青海大学生态环境工程学院,西宁 810016)
基于RAD-seq技术的异型花SSR信息分析
王久利1,2朱明星3徐明行3陈世龙1张发起1*
(1.中国科学院高原生物适应与进化重点实验室,中国科学院西北高原生物研究所,西北生态环境资源研究院,西宁 810001;2.中国科学院大学,北京 100039;3.青海大学生态环境工程学院,西宁 810016)
用RAD-seq(restriction-site associated DNA sequencing)对异型花(Sinoswertiatetraptera(Maxiowicz) T.N.Ho,S.W.Liu & J.Q.Liu)进行简化基因组测序,并借此分析异型花的SSR(simple sequence repeats)信息。利用SR search软件甄别所得序列中的SSR,得到了双端各有至少100 bp的SSR位点5 844个,其中5 339个(91.38%)成功设计引物,而三核苷酸SSR位点最多(3 323个);在能成功设计引物的SSR位点中,重复序列长度包括17种(12~36 bp);重复序列的基序共277种,其中五核苷酸基序种类最多(106种);随机挑选10对SSR引物,用4个异型花居群的32个个体检测检测其可用性和多态性,经PCR和聚丙烯酰氨凝胶电泳检测,有4对(ST2、ST3、ST6和ST10)成功扩增并表现出多态性;经GENEPOP 4.4对4个位点分析,显示其等位基因数量均值为6,多态性较高且不连锁(P<0.01);4个位点在多数居群中偏离哈迪温伯格平衡(P<0.01)且存在较高的纯合子数量(观测杂合度均值0.023),该结果归因于异型花主要进行自花授粉,在自然界中很难形成进行自由交配的居群;此外,ST2和ST6可在椭圆叶花锚(HaleniaellipticaD.Don)中成功扩增,具有潜在通用性。本研究将为日后基于异型花SSR标记的相关研究提供数据库支持。
RAD-seq;简化基因组测序;异型花;SSR;獐牙菜亚族
异型花(Sinoswertiatetraptera(Maxiowicz) T.N.Ho,S.W.Liu & J.Q.Liu)又名四数獐牙菜,是龙胆科(Gentianaceae)异型花属(Sinoswertia)的青藏高原特有的一年生草本植物[1~3]。特有种的DNA记录了其对特殊环境的响应等信息,这些信息对物种演化的研究具有重要意义[4~6];异型花也是一种重要的藏药,主要应用于肝胆疾病的治疗[7],随着近年来人们对传统医药的重视以及加工技术的发展,市场对传统药材的需求量增大,异型花也因此受到一定的威胁,因此有必要研究其DNA水平上的遗传多样性[8];目前对异型花的研究多集中在植物化学分析领域[8],然而在DNA水平上研究异型花遗传多样性的研究,已见报道的仅有Yang等[9]利用ISSR指纹图谱分析了异型花的居群遗传结构,并提出了对异型花的遗传多样性高的区域进行保护的保护策略。
简单重复序列(simple sequence repeats,SSR)又称微卫星(microsatellite),一般认为是由2至6个碱基对(base pair,bp)为重复单元的序列,大量分布于真核生物和原核生物基因组中。其共显性和高重复性比RAPD、ISSR和AFLP等分子标记更具优势[10],因而被用于指纹图谱、遗传变异分析、物种起源进化、基因定型、遗传图谱构建、品种评估和微生物鉴定等研究中[11~16]。
简化基因组测序(Reduced-representation sequencing)是一类利用不同方法得到目的基因组的部分区域,并对这些区域进行测序,从而反映基因组一部分序列结构信息的一种新兴的测序技术,其与全基因组测序相比具有低成本优势[17]。紧随技术的发展,简化基因组测序有产生了多种不同的方法,而利用限制性酶切位点相关的DNA测序(Restriction-site associated DNA sequence,RAD-seq)[18]是其中具有代表性的方法。RAD-seq具有通量高、准确性高、数据利用率高、实验周期短、不受基因组序列的限制以及性价比高等诸多优点[19],因而其广泛应用于分子标记开发、系统发育研究、种群结构分析、基因功能研究、物种起源研究等众多领域[20~23]。
为了在利用异型花资源的同时能更好地保护这一珍贵物种,本研究中将基于RAD-seq技术分析异型花的SSR信息并讨论基于RAD-seq技术开发异型花的多态性SSR标记的可行性,为异型花的SSR多态性位点引物的设计提供便利的数据库,进一步为基于异型花SSR多态性位点相关研究奠定基础。
1 材料与方法
1.1 样品的采集与DNA提取
本研究所用异型花样品分别采自定西、果洛、临夏和甘孜的4个自然居群,所用椭圆叶花锚的样品采自5个自然居群,各采样点的坐标和海拔信息在采集地中心使用GPS全球定位系统Etres GIS采集器记录(Garmin,中国台湾)(表1)。凭证标本保存于中国科学院西北高原生物研究所青藏高原植物标本馆(HNWP)。从每个居群随机选取的一个个体的叶片用于提取DNA,叶片的采集、处理的方法以及DNA提取的方法同Zhang等[24],并用1%的琼脂糖凝胶电泳和分光光度计NanoDrop 2000C(Thermo Scientific,USA)检测所提取的DNA,以保证DNA的总量和纯度。
1.2 文库构建与测序
将来自4个不同居群异型花的合格DNA样品等量混合之后,交由北京诺禾致源生物信息科技有限公司进行建库测序。建库过程具体参见Davey和Blaxter[25]。本研究回收300~700 bp的序列,通过HiSeq 2000基因分析系统(Illumina,USA)进行双向测序。
1.3 SSR位点甄别与引物设计
利用SR search软件(Novogene,中国北京)以重复单元长度2~6 bp、SSR最小长度12为标准,检测DNA序列所有SSR;过滤掉其中距离过近的SSR序列,两个SSR的最小距离为12 bp;为方便引物设计,SSR的重复序列的上下游各有长度为不短于100 bp的序列;然后利用SR search软件中的primer3模块设计SSR引物[26];参数设置:引物长度20~28 bp,最适长度24 bp,引物退火温度60~65℃且每对引物退火温度差最大值为1,最佳退火温度63℃。
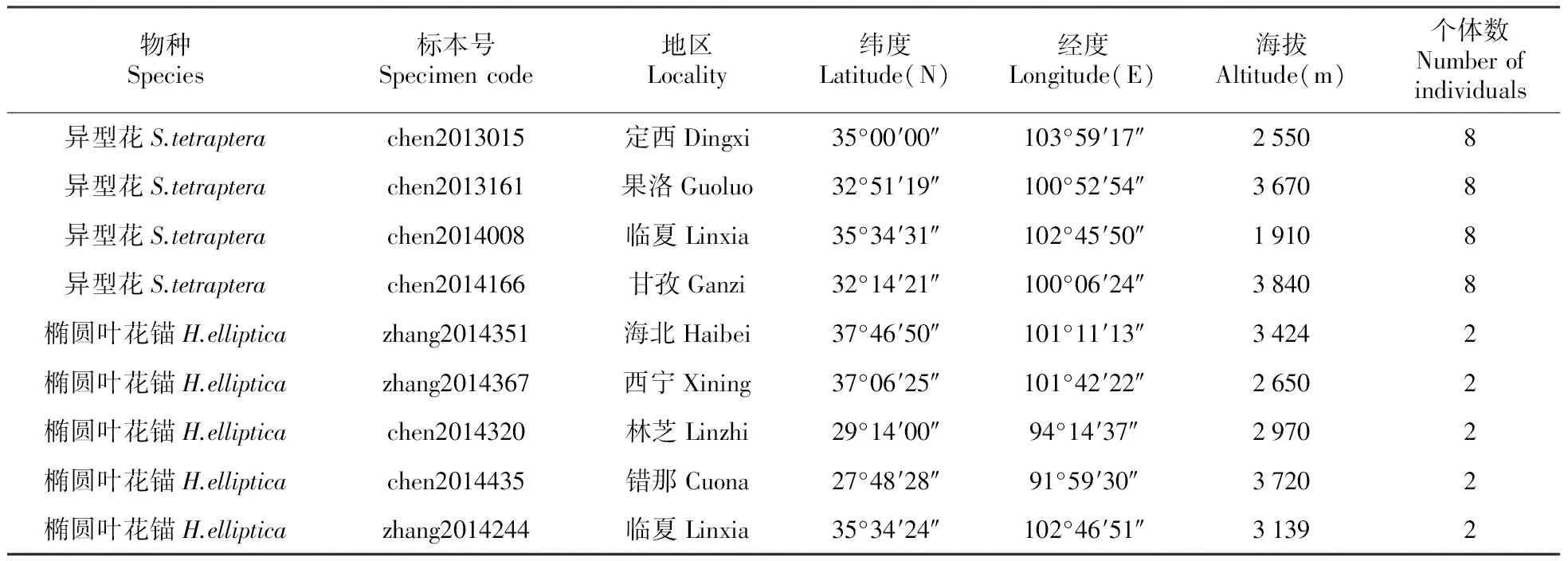
表1 异型花和椭圆叶花锚样品的居群信息
1.4 PCR扩增与检测
随机抽取10对SSR引物(对应的位点囊括二、三、四、五、六核苷酸重复类型,表4),利用梯度退火温度PCR反应选定引物的最佳退火温度:在20 μL的PCR反应总体积中,含30 ng·μL-1的DNA模板(4个来自不同居群的个体的混合DNA)0.9 μL,10×PCR Buffer 2.0 μL,10 μM正反向引物各0.4 μL,10 mmol·L-1dNTPs 0.7 μL,Taq DNA聚合酶(5 U·μL-1)0.1 μL,ddH2O 15.5 μL。PCR扩增在Mastercycler pro PCR仪(Eppendorf,德国)上进行,扩增程序为【95℃,5 min;(94℃,30 s;Tm,60 s;72℃,60 s)×25;72℃,7 min;16℃,∞】(Tm为各引物的退火温度)。接着,用1.5%的琼脂糖凝胶对PCR产物进行电泳检测,以溴化乙锭(ethidium bromide,EB)染色,在GelDoc 2000凝胶成像系统(Bio-Rad,美国)上拍照。根据琼脂糖凝胶电泳结果选定可成功PCR扩增的引物及其最佳退火温度。
然后用4个自然居群总共32个异型花个体(表1)对能成功扩增引物按照上述体系和程序进行PCR,其中退火温度换成相应引物的最佳退火温度。然后将PCR产物进一步用20%的聚丙烯酰氨凝胶进行电泳检测,然后同样以EB染色并在GelDoc 2000凝胶成像系统上拍照。
2 数据分析与结果
2.1 RAD-seq测序数据质量评估
异型花混池DNA的RAD测序共产生Raw data 2.76G,过滤后得到Clean data 2.71G。Phred数值大于20、30的碱基占总体碱基的百分比分别不低于93.35%和88.0%,GC含量为36.12%;RAD-Tag捕获率为97.68%。可见,样本的数据量足够,测序质量合格,GC分布正常,建库测序成功。
2.2 聚类
对样本含有酶识别位点reads用Cd-hit-est把RAD-tag相近的reads聚集到一个类中[27]。对每一类的reads支持数进行初步的过滤,过滤标准为每类中reads支持数为10~400,数据聚类后共得到1 275 817类,用于聚类的所有reads数为7 818 523,对reads支持深度进行过滤之后所剩下的pair reads数为5 127 265,过滤之后的reads数与所有参加聚类reads的比例(Cut/Pair)为65.58%。
2.3 组装
根据聚类结果用Velvetopt对筛选后的每一类进行局部组装[28],得到组装序列后过滤掉125 bp以下的contig。本次组装总长度为93 655 566 bp,组装总数为301 821,contig平均长度为310,序列从大到小排列,当长度达到组装总长度一半时,contig的长度N50=425 bp。组装结果中的GC含量为35.83%,而所有reads的GC含量为36.12%。
将去重复之后的reads通过Burrows-Wheeler alignment tool(BWA)软件比对到组装结果中[29],利用SAMtools检测变异情况[30]。检测结果显示,所有能比对上组装基因组的reads占所有参加比对reads的88.8%;每个位点深度为16.04;reads能够覆盖组装基因组的98.56%;reads能够覆盖组装基因组且深度不低于4×的比例是79.63%;自身reads比对组装结果所检测出的SNP数为252 614,其中杂合SNP为241 335,杂合比例为95.54%。样本比对率反映了样本Clean data的利用情况,覆盖深度和覆盖度足以直接反应测序数据的均一性及与组装基因组的一致性。
2.4 SSR位点及其引物
利用SR search软件对样本组装contig进行过滤,最终获得双端各有100 bp的SSR位点共5 844个,并成功设计5 339对SSR引物,成功设计率为91.38%。已成功设计引物的SSR位点的部分信息列于表2,我们获得的SSR最多的是三核苷酸(3 323条),其次是二核苷酸(1 167条),最少的是六核苷酸(89条);但是,重复序列的基序种类共277种,其中基序种类数最多的是五核苷酸(106种),其次是六核苷酸(89种),最少的是二核苷酸(11种)。从能成功设计引物的5 339个SSR位点的重复序列长度包括17种(12~36 bp),从其分布(表3)来看,长度在12 bp的SSR位点多达3 028个,占总SSR位点数的56.71%;其次是16 bp的SSR位点,为594个(11.13%);而不短于25 bp的SSR位点有127个占总数的12.38%(成功设计引物的5 339个SSR位点详细信息参见附件1)。
2.5 异型花SSR引物的有效性分析
通过梯度退火温度PCR从10对备选SSR引物中筛选出可在S.tetraptera中成功扩增的引物分别是ST2、ST3、ST6和ST10,并确定这4对引物的最佳退火温度。聚丙烯酰氨凝胶电泳显示这4对引物都表现出多态性,等位基因的大小变化范围见表4。
表2异型花SSR序列的信息
Table2InformationofSSRsequencesinS.tetraptera
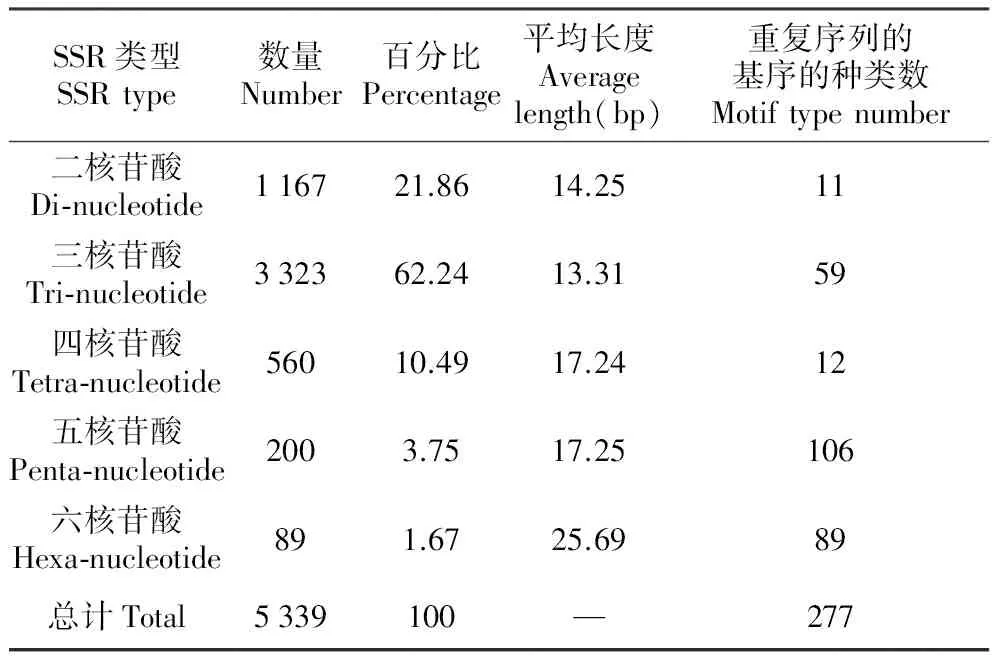
SSR类型SSRtype数量Number百分比Percentage平均长度Averagelength(bp)重复序列的基序的种类数Motiftypenumber二核苷酸Di⁃nucleotide116721.8614.2511三核苷酸Tri⁃nucleotide332362.2413.3159四核苷酸Tetra⁃nucleotide56010.4917.2412五核苷酸Penta⁃nucleotide2003.7517.25106六核苷酸Hexa⁃nucleotide891.6725.6989总计Total5339100—277
表3异型花SSR位点重复序列的长度分布
Table3DistributionofthelengthofSSRsequenceslociofS.tetraptera
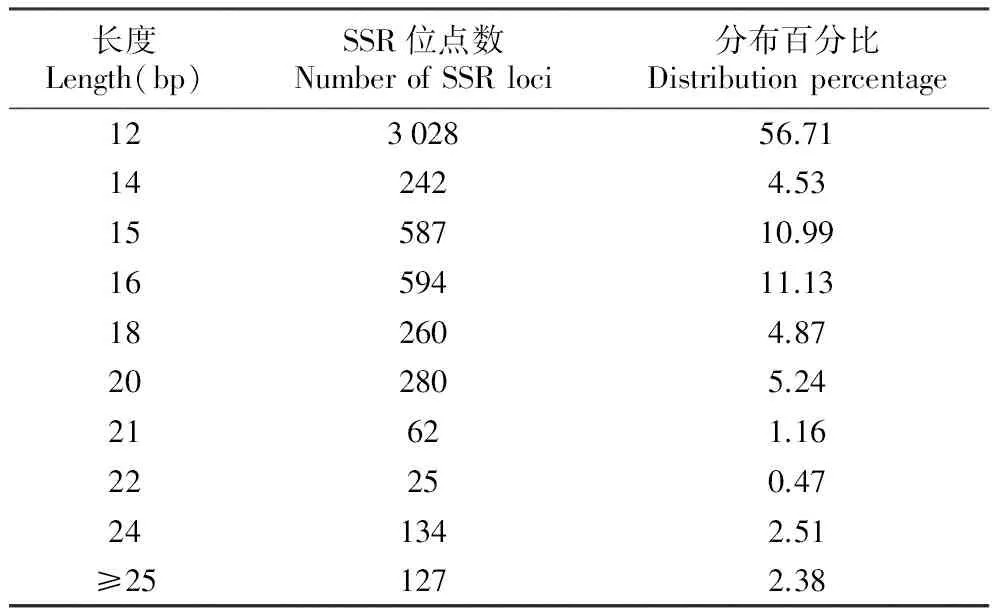
长度Length(bp)SSR位点数NumberofSSRloci分布百分比Distributionpercentage12302856.71142424.531558710.991659411.13182604.87202805.2421621.1622250.47241342.51≥251272.38
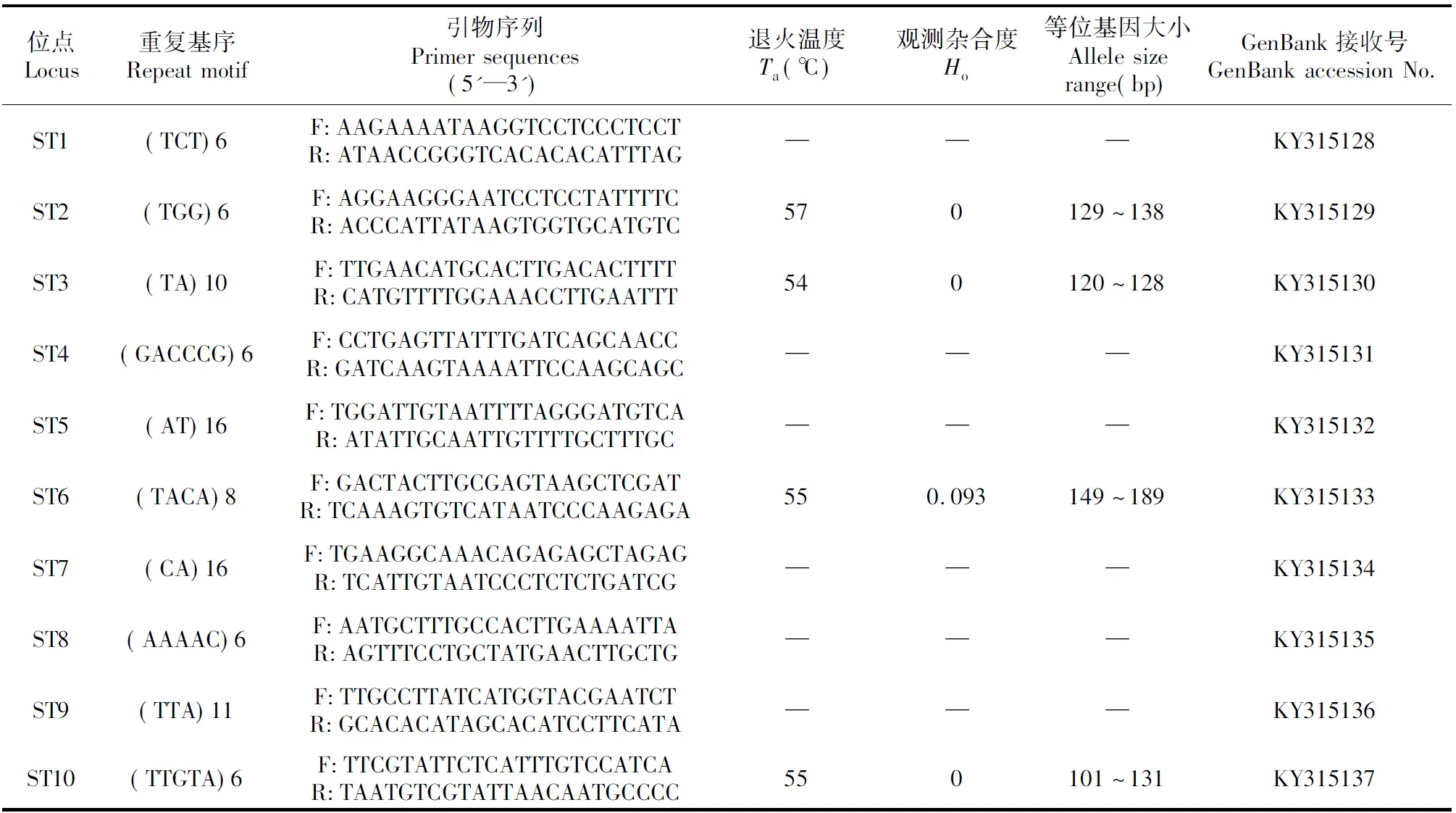
表4 异型花的10对引物特点
利用GENEPOP 4.4[31]分析聚丙烯酰氨凝胶电泳结果。所检测的4对引物所对应的位点的等位基因数为4~9,均值为6,多态性较高;连锁不平衡(Linkage Disequilibrium)分析显示4个SSR位点不连锁(P<0.01);而ST2、ST3、ST6和ST10的观测杂合度分别为0、0、0.093和0,均值0.023。
异型花属是单型属,其近缘类群为花锚属(HaleniaBorkh.)和獐牙菜属(SwertiaLinn.)[2~3],因而用椭圆叶花锚(HaleniaellipticaD.Don)对异型花多态性引物进行近缘物种交叉检验,发现ST2和ST6可以在椭圆叶花锚中成功扩增。
3 讨论
目前,异型花尚未获得全基因组或转录组数据,甚至也没有龙胆科内近缘物种的可参考全基因组序列。因而我们采用RAD-seq技术对异型花进行简化基因组测序,测序结果成功,获得了异型花基因组水平上的序列信息。所得的序列经聚类和组装后,也呈较高的组装准确性,从侧面反映组装结果能够代表部分基因组。样本比对率(88.80%)反映了样本Clean data的高利用率,覆盖深度和覆盖度足以直接反映测序数据的均一性及与组装基因组的一致性。
利用SR search软件对获得的序列进行SSR甄别,获得双端各有100 bp的SSR位点多达5 844个,并成功设计5 339对SSR引物,成功设计率高达91.38%。在能成功设计引物的SSR位点中,重复序列的基序种类共277种,而且重复序列长度包括17种,可见已成功设计引物的异型花的SSR多样性较高。随机挑选的10对SSR引物中,经PCR和凝胶电泳检测,有4对可在S.tetraptera中成功扩增,并且表现出多态性。经GENEPOP 4.4对已获得的位点分析,显示其等位基因的均值为6,多态性较高,并且相互之间不连锁;4个位点偏离哈迪温伯格平衡,这是因为异型花主要进行自花授粉,在自然界中很难形成进行自由交配的居群[2~3],较高的纯合子数量(观测杂合度均值0.023)可能也归因于其授粉方式;4对引物中有2对可以在其近缘异属中成功扩增,表现出了一定的通用性。可见,本研究将为后续的基于SSR标记的异型花居群遗传和指纹图谱等研究提供足够的SSR标记数据库。
值得补充的是,我们用同样的方法开发了肉果草(LanceatibeticaHook.F. et Thoms)[23]和椭圆叶花锚(待发表数据)的多态性SSR位点,通过与我们以往的窄叶鲜卑花(Sibiraeaangustata(Rehder) K.S.Hao)[32]、绣线菊属(SpiraeaLinn.)[33]、西川红景天(Rhodiolaalsia(Frod.) S.H.Fu)[34]和黄绿蜜环菌(Armillarialuteo-virens(Aalb.et Schw:Fr.)Sacc.)[35]等物种的SSR标记开发研究的比较,我们发现基于RAD-seq技术进行SSR标记研究表现出了RAD-seq技术本身所具备的优势,包括通量高、准确性高、数据利用率高、实验周期短、不受基因组序列的限制以及性价比高[18~19],在SSR位点开发方面明显优于传统的FIASCO(Fast Isolation by AFLP of Sequences Containing Repeats)[36]等方法。所以,对于尚无足够核酸序列数据参考的物种,RAD-seq是目前的一种比较理想的SSR信息研究技术。
1.He T N,Liu S W,Chen S L.Nomenclatural novelities inSwertia(Gentianaceae)[J].Plant Diversity and Resources,2013,35(3):386-392.
2.He T N,Liu S W,Liu J Q.A new Qinghai-Tibet Plateau endemic genusSinoswertiaand its pollination mode[J].Plant Diversity and Resources,2013,35(3):393-400.
3.Ho T N,Liu S W.A worldwide monograph ofSwertiaand its allies[M].Beijing:Science Press,2015:301-308.
4.Fu P C,Gao Q B,Zhang F Q,et al.Responses of plants to changes in Qinghai-Tibetan Plateau and glaciations:Evidence from phylogeography of aSibiraea(Rosaceae) complex[J].Biochemical Systematics and Ecology,2016,65:72-82.
5.Gao Q B,Zhang D J,Duan Y Z,et al.Intraspecific divergences ofRhodiolaalsia(Crassulaceae) based on plastid DNA and internal transcribed spacer fragments[J].Botanical Journal of the Linnean Society,2012,168(2):204-215.
6.Wang L Y,Abbott R J,Zhang W,et al.History and evolution of alpine plants endemic to the Qinghai-Tibetan Plateau:Aconitumgymnandrum(Ranunculaceae)[J].Molecular Ecology,2009,18(4):709-721.
7.中国科学院西北高原生物研究所.藏药志[M].青海:青海人民出版社,1991:111-112.
Northwest Institute of Plateau Biology,Chinese Academy of Sciences.Handbook of Tibetan medicine[M].Qinhai:Qinghai People’s Publishing Press,1991:111-112.
8.杨路存,陈桂琛,周国英,等.四数獐牙菜ISSR-PCR反应体系的正交优化[J].生物技术通报,2010,20(9):123-128.
Yang L C,Chen G C,Zhou G Y,et al.Optimization for ISSR-PCR reaction system inSwertiatetrapterausing orthogonal design[J].Biotechnology Bulletin,2010,20(9):123-128.
9.Yang L C,Zhou G Y,Chen G C.Genetic diversity and population structure ofSwertiatetraptera(Gentianaceae),an endemic species of Qinghai-Tibetan Plateau[J].Biochemical Systematics and Ecology,2011,39(4-6):302-308.
10.Wang X R,Szmidt A E.Molecular markers in population genetics of forest trees[J].Scandinavian Journal of Forest Research,2001,16(3):199-220.
11.何平.真核生物中的微卫星及其应用[J].遗传,1998,20(4):42-47.
He P.Abundance,Polymorphism and applications of microsatellite in eukaryote[J].Hereditas,1998,20(4):42-47.
12.Doerge R W.Mapping and analysis of quantitative trait loci in experimental populations[J].Nature Reviews Genetics,2002,3(1):43-52.
13.Liu W J,Nie H,Wang S B,et al.Mapping a resistance gene in wheat cultivar Yangfu 9311 to yellow mosaic virus,using microsatellite markers[J].Theoretical and Applied Genetics,2005,111(4):651-657.
14.Yoshimoto K,Yoshida J,Ishii G,et al.Two lung adenocarcinomas in the same lobe:multiple primaries or intrapulmonary metastasis?[J].Annals of Thoracic and Cardiovascular Surgery,2011,17(6):584-587.
15.陶星辰,尚秋菊,刘晓英,等.云南省部分地区鸽粪中新型隐球菌的分离与鉴定[J].中国病原生物学杂志,2014,9(10):873-876.
Tao X C,Shang Q J,Liu X Y,et al.Isolation and identification ofCryptococcusneoformansfrom pigeon droppings in parts of Yunnan Province[J].Journal of Pathogen Biology,2014,9(10):873-876.
16.Conti S,Gallo E,Sioletic S,et al.Molecular genetic alterations in egfr CA-SSR-1 microsatellite and egfr copy number changes are associated with aggressiveness in thymoma[J].Journal of Thoracic Disease,2016,8(3):386-395.
17.石璇,王茹媛,唐君,等.利用简化基因组技术分析甘薯种间单核苷酸多态性[J].作物学报,42(5):641-647.
Shi X,Wang R Y,Tang J,et al.Analysis of interspecific SNPs in sweetpotato using a Reduced-Representation genotyping technology[J].Acta Agronomica Sinica,2016,42(5):641-647.
18.Miller M R,Dunham J P,Amores A,et al.Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA(RAD) markers[J].Genome Research,2016,17(2):240-248.
19.王洋坤,胡艳,张天真.RAD-seq技术在基因组研究中的现状及展望[J].遗传,2014,36(1):41-49.
Wang Y K,Hu Y,Zhang T Z.Current status and perspective of RAD-seq in genomic research[J].Hereditas,2014,36(1):41-49.
20.Willing E M,Hoffmann M,Klein J D,et al.Paired-end RAD-seq for de novo assembly and marker design without available reference[J].Bioinformatics,2011,27(16):2187-2193.
21.Hou Y,Nowak M D,Mirr V,et al.RAD-seq data point to a northern origin of the arctic-alpine genusCassiope(Ericaceae)[J].Molecular Phylogenetics and Evolution,2016,95:152-160.
22.Gamble T,Coryell J,Ezaz T,et al.Data from:Restriction site-associated DNA sequencing(RAD-seq) reveals an extraordinary number of transitions among gecko sex-determining systems[J].Molecular Biology and Evolution,2015,32(5):1296-1309.
23.Tian Z Z,Zhang F Q,Liu H R,et al.Development of SSR markers for a Tibetan medicinal plant,Lanceatibetica(Phrymaceae),based on RAD sequencing[J].Applications in Plant Sciences,2016,4(11):1600076.
24.Zhang F Q,Lei S Y,Gao Q B,et al.Isolation of microsatellite loci forRhodiolaalsia(Crassulaceae):an important ethno-medicinal herb endemic to the Qinghai-Tibetan plateau[J].Genetics and Molecular Research,2015,14(2):5266-5269.
25.Davey J W,Blaxter M L.RADSeq:next-generation population genetics[J].Briefings in functional genomic,2010,9(5-6):416-423.
26.Rozen S,Skaletsky H.Primer3 on the WWW for general users and for biologist programmers[M].//Misener S,Krawetz S A,eds.Bioinformatics Methods and Protocols.Totowa,NJ:Humana Press,1999:365-386.
27.Li W Z,Godzik A.Cd-hit:a fast program for clustering and comparing large sets of protein or nucleotide sequences[J].Bioinformatics,2006,22(13):1658-1659.
28.Zerbino D R,Birney E.Velvet:algorithms for de novo short read assembly using de Bruijn graphs[J].Genome Research,2008,18(5):821-829.
29.Li H,Durbin R.Fast and accurate long-read alignment with burrows-wheeler transform[J].Bioinformatics,2010,25(5):589-595.
30.Li H,Handsaker B,Wysoker A,et al.The sequence alignment/map format and SAMtools[J].Bioinformatics,2009,25(16):2078-2079.
31.Rousset F.GENEPOP’007:A complete re-implementation of the GENEPOP software for Windows and Linux[J].Molecular Ecology Resources,2008,8(1):103-106.
32.Arranz S E,Avarre J C,Balasundaram C,et al.Permanent Genetic Resources added to Molecular Ecology Resources Database 1 December 2012-31 January 2013[J].Molecular Ecology Resources,2013,13(3):546-549.
33.张金华,Khan G,付鹏程,等.基于磁珠富集法的绣线菊SSR分子标记分离与筛选[J].生物学杂志,2014,31(3):79-83.
Zhang J H,Khan G,Fu P C,et al.Isolation and screening of molecular genetic marker SSR inSpiraeabased on magnetic beads enriched[J].Journal of Biology,2014,31(3):79-83.
34.王久利,雷淑芸,陈世龙,等.基于磁珠富集法开发并筛选西川红景天的微卫星标记[J].广西植物,2017(03):342-347.
Wang J L,Lei S Y,Chen S L,et al.Development and screening of SSR markers inRhodiolaalsiabase on magnetic beads method[J].Guihaia,2017(03):342-347.
35.邢睿,高庆波,张发起,等.基于454测序技术的青藏高原黄绿蜜环菌微卫星引物的开发[J].微生物学报,2014,54(9):1045-1052.
Xing R,Gao Q B,Zhang F Q,et al.Isolation and characterization of 27 polymorphic microsatellite markers inArmillarialuteo-virens(Physalacriaceae)[J].Acta Microbiologica Sinica,2014,54(9):1045-1052.
36.Zane L,Bargelloni L,Patarnello T.Strategies for microsatellite isolation:a review[J].Molecular Ecology,2002,11(1):1-16.
National Natural Science Foundation of China(31400322);Applied Basic Research Programs of Qinghai Province(2016-ZJ-761)
introduction:WANG Jiu-Li(1991—),male,doctoral candidate,Botany major.
date:2016-12-26
AnalysisonSSRinSinoswertiatetrapteraBaseonRAD-seq
WANG Jiu-Li1,2ZHU Ming-Xing3XU Ming-Hang3CHEN Shi-Long1ZHANG Fa-Qi1*
(1.Key Laboratory of Adaptation and Evolution of Plateau Biota,Northwest Institute of Plateau Biology,Northwest Institute of Eco-Environment and Resources,Chinese Academy of Sciences,Xining 810008;2.University of the Chinese Academy of Sciences,Beijing 100039;3.College of Ecology-Environment Engineering,Qinghai University,Xining 810016)
We used the restriction-site associated DNA sequencing(RAD-seq) technology to analyze simple sequence repeats(SSR) information ofSinoswertiatetraptera(Maxiowicz) T.N.Ho, S.W.Liu & J.Q.Liu. The 5844 SSR loci, with at least 100 bp at two ends, were identified using SR search software. The 5339(91.38%) loci’s primers were designed successfully. Among which, amount of tri-nucleotide SSR loci is the most(3323); repeat sequence length type number is 17, while repeat motif type number is 227, and type number of penta-nucleotide motif is the most(106). We employed 32 individuals from 4 natural populations ofS.tetrapterato estimate usability and polymorphism of 10 pairs of SSR primers selected randomly from the 5 339 pairs of primers. According to the result of PCR and Polyacrylamide gel electrophoresis, four pairs of primers(ST2, ST3, ST6 and ST10) amplified favorably and showed polymorphism. By the GENEPOP 4.4, the mean number of alleles of the four loci is 6; these loci do not link to each other(P<0.01); these loci deviate from HWE(P<0.01) in most populations and have many homozygotes(observed heterozygosity mean 0.023), which due to the cleistogamous pollination mode ofS.tetraptera. ST2 and ST6 were successfully amplified inHaleniaelliptica. Our study will offer a SSR dataset in the future based on SSR markers ofS.tetraptera.
RAD-seq;reduced-representation sequencing;Sinoswertiatetraptera;SSR;subtribe Swertiinae
国家自然科学基金(31400322);青海省应用基础研究计划(2016-ZJ-761)
王久利(1991—),男,博士研究生,主要从事植物学方面的研究。
* 通信作者:E-mail:fqzhang@nwipb.cas.cn
2016-12-26
* Corresponding author:E-mail:fqzhang@nwipb.cas.cn
Q949.776.4
A
10.7525/j.issn.1673-5102.2017.03.016
