基于MiSeq高通量测序方法研究酵母壁多糖对断奶仔猪盲肠菌群结构的影响
2017-09-20王自蕊游金明陈丽玲卢亚飞李兰海
贺 琴,王自蕊,游金明⋆,陈丽玲,2,卢亚飞,李兰海
(1.江西农业大学动物科技学院,江西省动物营养重点实验室,江西省营养饲料开发工程中心,江西南昌330045;2.江西中医药大学实验动物科技中心,江西南昌 330004)
基于MiSeq高通量测序方法研究酵母壁多糖对断奶仔猪盲肠菌群结构的影响
贺 琴1,王自蕊1,游金明1⋆,陈丽玲1,2,卢亚飞1,李兰海1
(1.江西农业大学动物科技学院,江西省动物营养重点实验室,江西省营养饲料开发工程中心,江西南昌330045;2.江西中医药大学实验动物科技中心,江西南昌 330004)
本试验旨在探讨饲粮中添加酵母壁多糖对断奶仔猪盲肠菌群结构的影响。采用单因素试验设计,选取180头遗传背景相同、胎次相近、体重接近的21日龄断奶仔猪,随机分为4个处理,每个处理5个重复,每个重复9头猪。4个处理分别饲喂0 %酵母壁多糖饲粮(对照组)、0.15%酵母壁多糖饲粮(0.15%组)、0.30%酵母壁多糖饲粮(0.30%组)和0.45%酵母壁多糖饲粮(0.45%组)。试验期为21 d。取盲肠内容物提取细菌DNA,PCR扩增获得16S rRNA基因V4标签片段,进行MiSeq高通量测序。结果表明:当酵母壁多糖添加量达一定程度时,仔猪盲肠菌群多样性会发生改变;在不考虑群落中每个物种丰度的前提下,0.30%酵母壁多糖组仔猪盲肠菌群最丰富;仔猪盲肠优势菌群为拟杆菌门、厚壁菌门、螺旋体门和变形菌门;与对照组相比,添加0.30%酵母壁多糖能降低仔猪肠道拟杆菌门含量(P<0.05),添加0.30%或0.45%酵母壁多糖能显著增加盲肠厚壁菌门含量(P<0.05),添加0.45%酵母壁多糖还能提高仔猪盲肠软壁菌门含量(P<0.05);酵母壁多糖具有降低盲肠变形菌门含量的趋势(P=0.066);普氏菌属为仔猪盲肠优势菌属,酵母壁多糖能显著提高仔猪盲肠厌氧弧菌属、瘤胃杆菌属、粪球菌属和考拉杆菌属的含量(P<0.05),显著降低链球菌属的含量(P<0.05)。由此可知,当酵母壁多糖添加量达到一定水平时,短期添加可提高断奶仔猪盲肠厚壁菌门以及瘤胃杆菌属和粪球菌属等优势菌群的含量。
酵母壁多糖;断奶仔猪;菌群结构;菌群多样性
肠道微生态是哺乳动物体内最复杂的微生态系统。微生物种类多达1 000~1 150种,且细菌总数是宿主细胞数量的10倍[1-2]。在正常情况下,机体胃肠道微生物处于稳定的平衡状态。但是畜禽的日龄、遗传背景和饲养环境会改变肠道菌群结构[3]。在外界环境发生变化或饲粮改变等应激状态下,肠道微生物区系平衡会发生改变,可能导致机体肠道有害菌增殖,使得宿主健康受到影响。受饲粮等因素影响,断奶期是仔猪肠道菌群最不稳定的时期。稳定的肠道菌群能够保证胃肠道正常的消化和吸收。同时,在胃肠道定植的有益菌群可以形成机体内非特异性生物保护屏障,一定程度上可抑制病原菌定殖。因此,动物胃肠道微生物平衡是畜禽健康生长和正常生产的必要前提[4]。益生元是一种不可消化的饲粮添加剂,它能选择性地刺激肠道中潜在的有益微生物,并减少病原微生物的生长,从而改变肠道微生物群的组成并改善宿主健康[5-6]。早前研究发现,酵母壁多糖能改善断奶仔猪的生长性能,提高肠道挥发性脂肪酸含量,并可降低肠道大肠杆菌和沙门氏菌数量[7-8]。但受传统微生物研究方法限制,酵母壁多糖对肠道微生物菌群影响的研究甚少。目前,核酸测序技术能够更全面、准确地反映肠道菌群的结构与组成,因此本研究基于MiSeq高通量测序方法,测定短期饲喂酵母壁多糖对断奶仔猪盲肠菌群结构的影响,以期为酵母壁多糖在生产实践中的应用提供理论依据。
1 材料与方法
1.1 试验材料 酵母壁多糖为啤酒酵母壁多糖,来源于拓普生物科技有限公司,其主要成分为甘露寡糖(23.45%)、β-葡聚糖(39.24%)。粗蛋白质含量为26.30%、粗灰分含量为3.30%、水分含量为5.35%。
1.2 试验设计和饲养管理 采用单因素试验设计,选取180头遗传背景相同、胎次相近、体重接近的21日龄断奶仔猪,随机分为4个处理,每个处理5个重复,每个重复9头仔猪。4个处理分别饲喂对照饲粮(对照组)、0.15%酵母壁多糖饲粮(0.15%组)、0.45%酵母壁多糖饲粮(0.30%组)和0.45%酵母壁多糖饲粮(0.45%组)。试验期为21 d。试验开始前对猪舍进行充分冲洗和严格消毒。试验期内仔猪饲养于装有高床、漏缝地板、乳头式饮水器的保育舍。每日饲喂仔猪4~5次,所有仔猪自由采食和饮水,其他饲养管理措施、免疫程序按猪场常规管理程序进行。饲粮配方参照NRC(2012)和我国猪饲养标准(2004)配制,饲粮组成和营养成分见表1。
1.3 样品采集及处理 在试验第21天,从每个重复中选取1头接近平均体重且健康状况良好的仔猪,肌注4%戊巴比妥钠溶液进行麻醉。待麻醉完全后,采用颈静脉放血将其处死,在无菌状态下采集盲肠内容物,置于液氮中速冻,-80℃中保存。
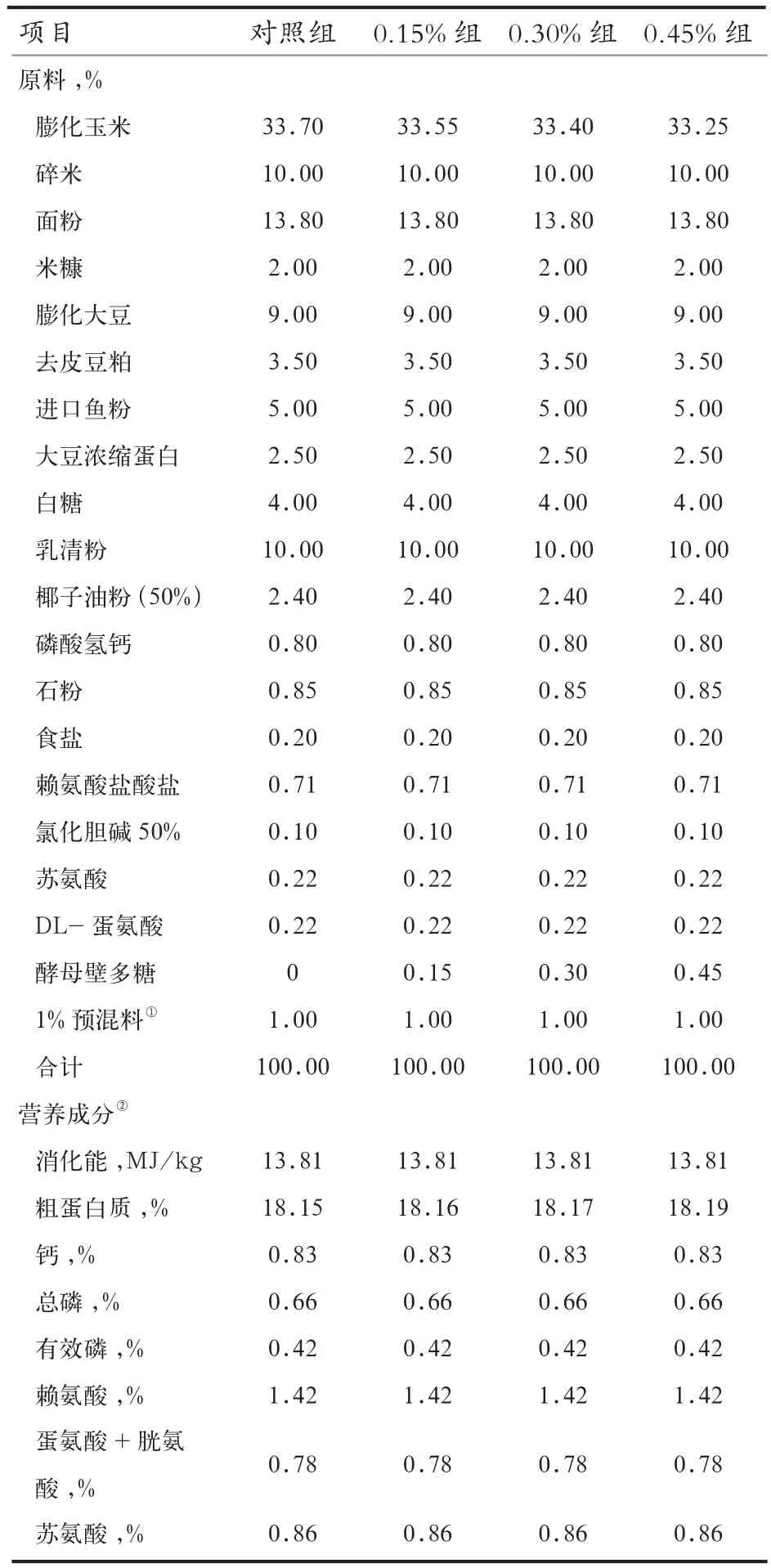
表1 试验饲粮组成及营养成分(风干基础)
1.4 盲肠微生物分析
1.4.1 引物的设计 利用细菌总DNA作为模板,使用细菌16S rRNA基因V4区通用引物,进行PCR扩增。其中通用引物序列:515F:5'-GTGCCAGCMGCCGCGGTAA-3',806R:5'-GGACTACHVGGGTWTCTAAT-3'。
1.4.2 盲肠总DNA提取和MiSeq测序 将盲肠样品4℃条件下解冻,采用QIAamp DNA Stool MiniKit提取微生物总DNA,使用1%浓度的琼脂糖凝胶在150 V电压下电泳约40 min,利用NanoDrop®ND1000测定DNA条带质量。纯化后的16S r RNA基因V4区的PCR产物送至华大基因检测,通过Illumina MiSeq平台进行Paired-end测序。
1.4.3 生物分析流程 对下机数据进行质量控制,舍弃低质量序列,获得的高质量Clean Data用于后期分析。通过reads之间的Overlap关系将reads拼接成Tags。以16S rDNA序列97%的相似度作为分类操作单元(Operational Taxonomic Unit,OTU)的划分标准。通过OTU与数据库比对,对OTU进行物种注释。利用mothur对获得的OTU做rarefaction、丰富度(Chao和Ace)和多样性分析(Shannon和Simpson),并做组间物种差异分析。
1.5 统计分析 所有数据用Excel 2003简单处理后,采用SPSS 17.0软件One-Way ANOVA模型进行方差分析数据以平均数±标准差表示。以P<0.05为显著性判断标准。
2 结果与分析
2.1 Alpha多样性分析 对20个样品进行测序,通过reads之间的overlap关系拼接成Tags,一共得到619 399条Tags,平均每个样品30 969条。对细菌进行Rank-Abundance分析(图1),从横轴上的长度来看,物种组成丰富度、物种组成均匀度由高到低均为0.30%酵母壁多糖组、0.15%酵母壁多糖组、0.45%酵母壁多糖组、对照组。
由图2可知,各组细菌Alpha指数稀释曲线在测序条数达到15 000条以上时趋于平台期。由表2可知,各组Sobs指数、Chao指数和Ace指数虽然差异不显著(P>0.05),但随酵母壁多糖添加量增多,Chao指数和Ace指数呈现先增加后降低的趋势。在不考虑群落中每个物种的丰度前提下,0.30%酵母壁多糖组仔猪盲肠菌群最丰富。Shannon指数和Simpson指数在各组间差异不显著(P>0.05)。
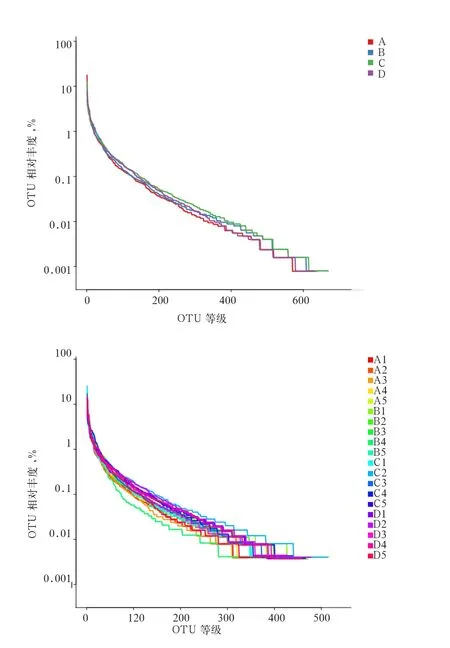
图1 酵母壁多糖对断奶仔猪盲肠细菌影响的Rank-Abundance分析图
2.2 Beta多样性分析 为了进一步研究样品多样性方面存在的差异,对样品进行Beta多样性(Beta Diversity)分析。基于样品OTU进行聚类分析,在考虑了序列丰度的前提下,构建盲肠细菌多样系相似度树状图。由图3可知,各组重复性较好,且随酵母壁多糖添加量提高,各组仔猪盲肠内容物菌群相似度越来越低,0.45%酵母壁多糖与对照组相比相似度最低。
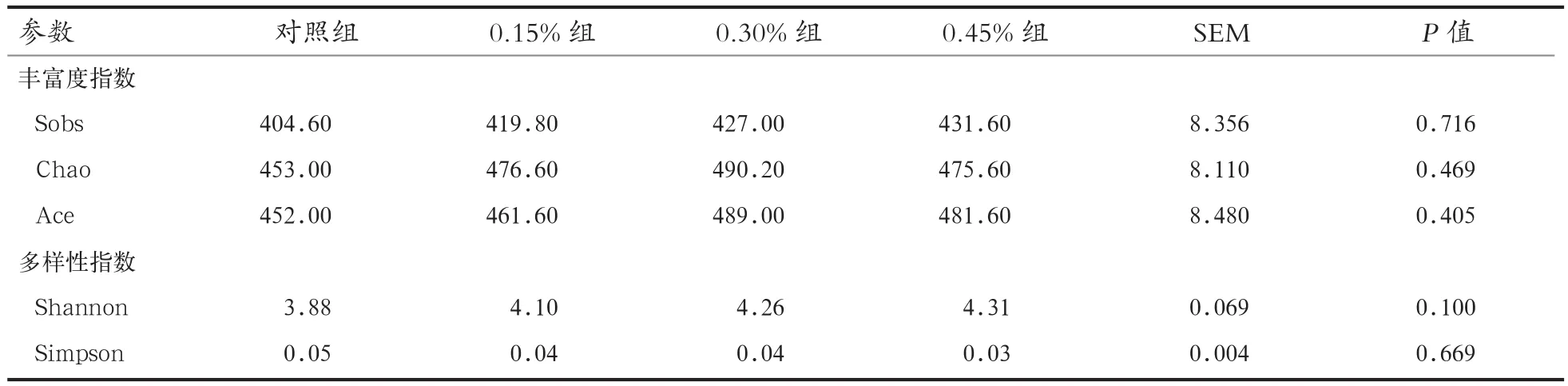
表2 酵母壁多糖对断奶仔猪盲肠细菌群落α-多样性影响
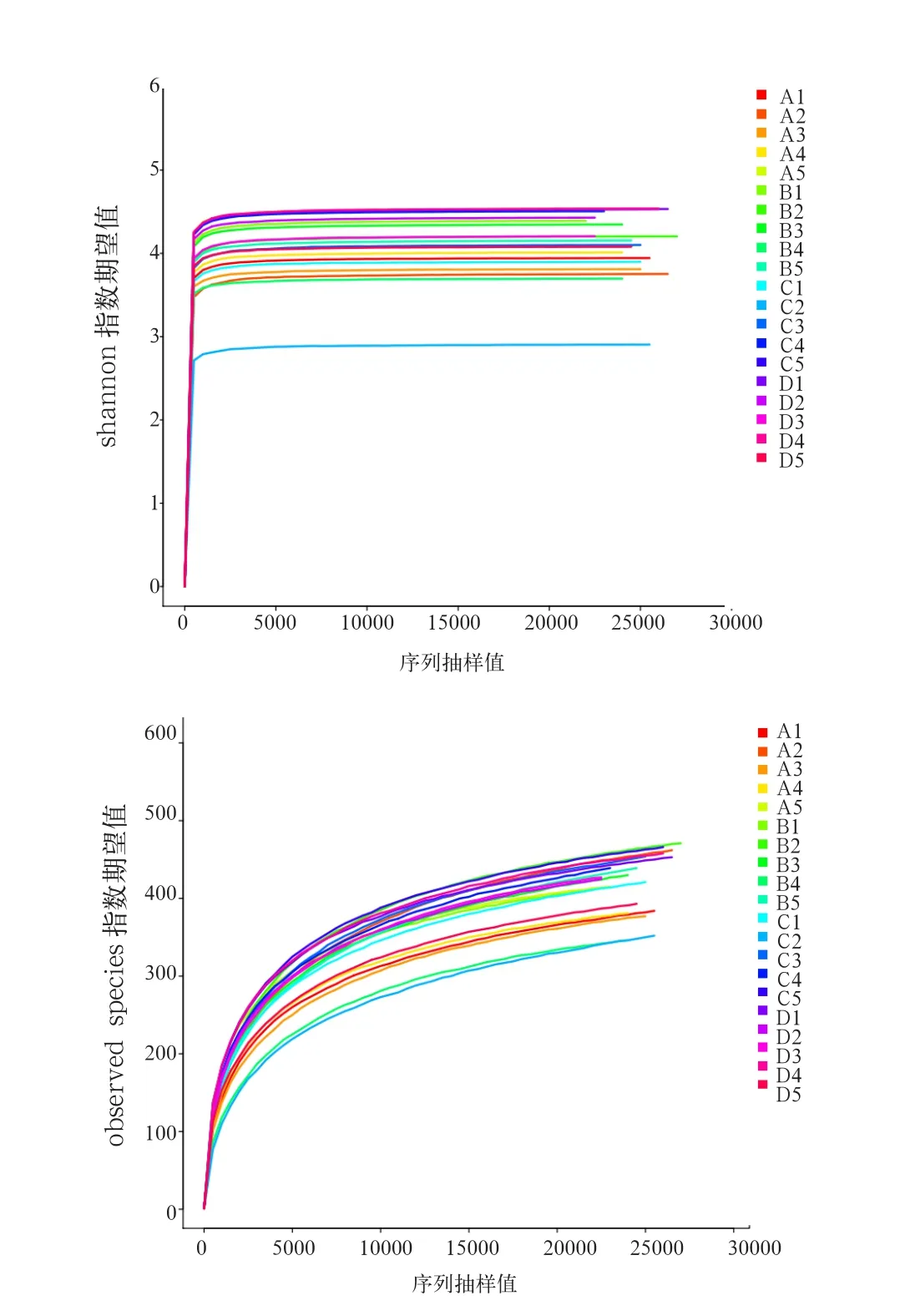
图2 酵母壁多糖对断奶仔猪盲肠细菌群落多样性Alpha指数稀释曲线影响
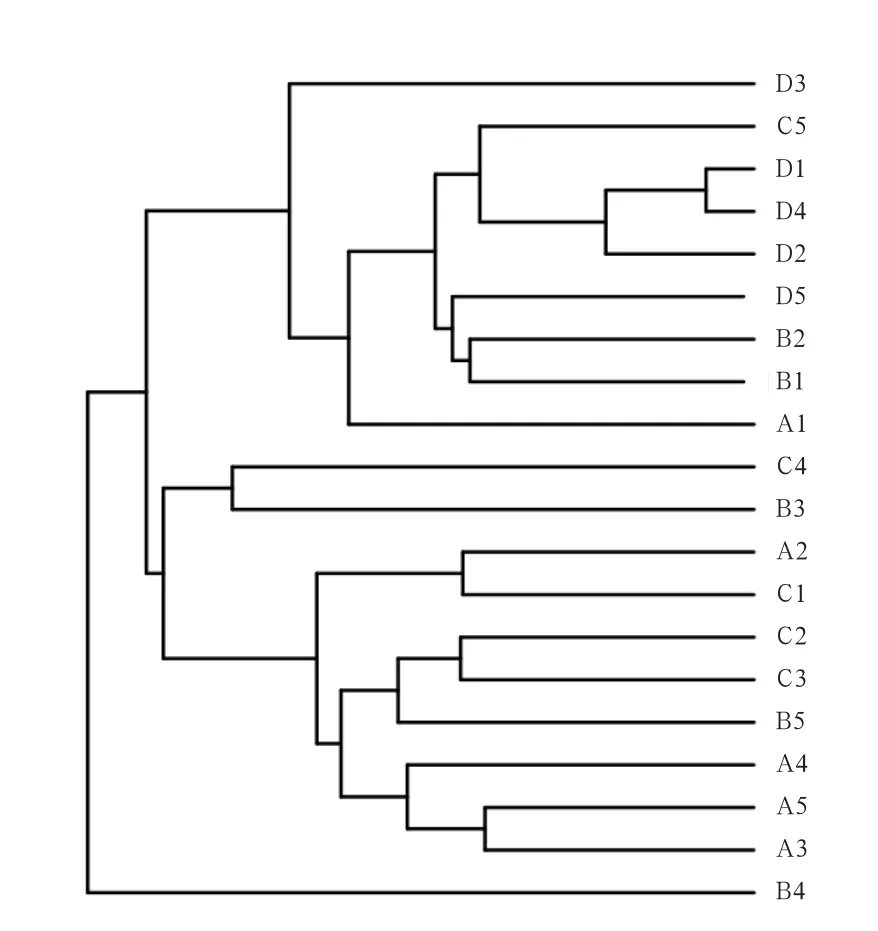
图3 酵母壁多糖对断奶仔猪盲肠细菌影响的聚类分析树状图(Weighted UniFrac)
2.3 菌群结构分析 对照组、0.15%酵母壁多糖组、0.30%酵母壁多糖组和0.45%酵母壁多糖组OTUs总数分别为660、699、682和686。共有的OTU数为533。对OTU进行物种分析,16个门、26个纲、61个科、81个属和102个种在所有样品中被鉴定。由表3可知,Bacteroidetes(拟杆菌门)、Firmicutes(厚壁菌门)、Spirochaetes(螺旋体门)和Proteobacteria(变形菌门)相对含量较高;Tenericutes(软壁菌门)、Deferribacteres(脱铁杆菌门)、Actinobacteria(放线菌门)和Fibrobacteres(纤维杆菌门)含量较低,属于劣势菌门;拟杆菌门、厚壁菌门和软壁菌门在各组间差异显著,与对照组相比,添加0.30%酵母壁多糖能显著降低仔猪肠道拟杆菌门含量(P<0.05),而添加0.30%和0.45%酵母壁多糖显著增加盲肠厚壁菌门含量(P<0.05);添加0.45%酵母壁多糖还能显著提高仔猪盲肠软壁菌门含量(P<0.05)。此外,酵母壁多糖具有降低盲肠变形菌门含量的趋势(P=0.066)。
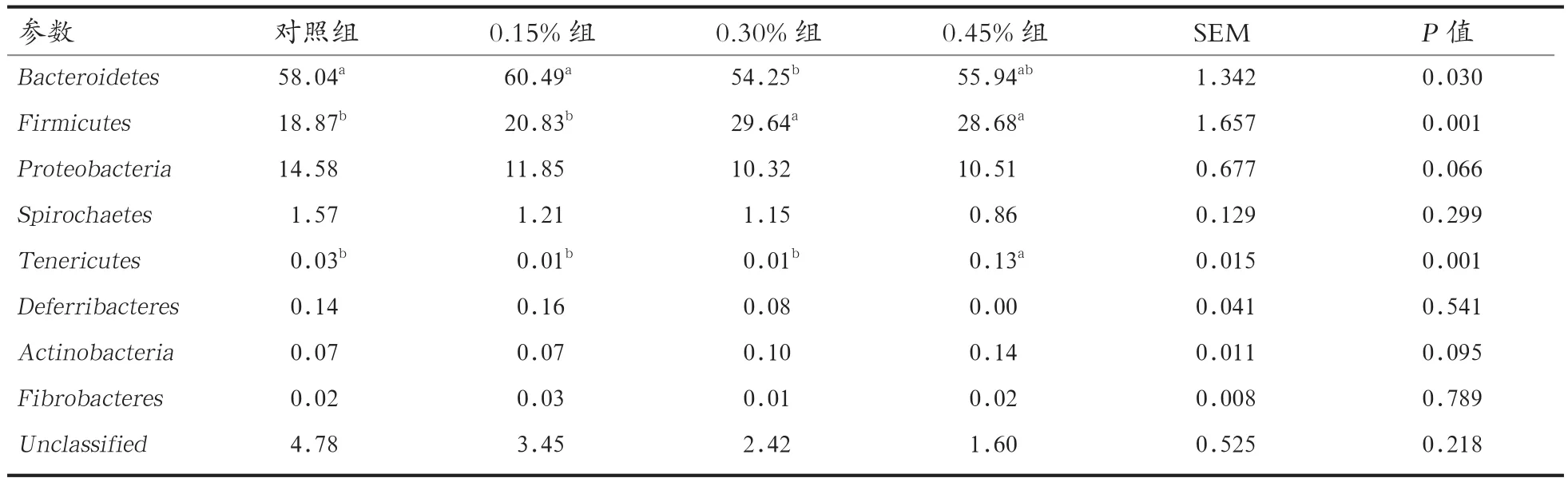
表3 不同水平酵母壁多糖对盲肠细菌组成的影响(门水平)
由表4可知,Prevotella(普氏菌属)、Bacteroides(拟杆菌属)、Oscillospira(颤螺菌属)、Parabacteroides(紫单胞菌属)、Phascolarctobacterium(考拉杆菌属)、Sphaerochaeta属和CF231属是仔猪盲肠优势菌属。添加0.30%和0.45%酵母壁多糖能提高仔猪盲肠Anaerovibrio(厌氧弧菌属)和Ruminococcus(瘤胃杆菌属)含量(P<0.05),并降低Streptococcus(链球菌属)的含量(P<0.05)。而相比于对照组,添加0.30%酵母壁多糖能提高盲肠CF231属含量(P<0.05)。此外,酵母壁多糖还能提高仔猪盲肠Coprococcus(粪球菌属)和Phascolarctobacterium(考拉杆菌属)含量(P<0.05),并降低Sphaerochaeta属含量(P<0.05)。
3 讨 论
后肠段是仔猪肠道中微生物的丰富部位,其中盲肠微生物菌群多样性较高,数量达每克食糜1011个[9]。肠道内的菌群粘附于肠黏膜上,并与肠上皮细胞粘连,形成机体的微生物屏障。它不仅改变了肠道的通透性,还与机体对营养物质的吸收、代谢和免疫系统有着不可分割的联系。在某种程度上,可将肠道菌群视为一种特殊的机体器官[1]。肠道微生物的动态平衡是仔猪健康生长的重要前提。在肠道菌群平衡的基础上,肠道中有益菌含量越高,畜禽越健康[10]。环境和日粮因素均会改变肠道菌群。
OTU Rank曲线是展现样品中物种多样性的一种形式,可以同时解释样品所含物种的丰富程度和均匀程度。Alpha多样性(Alpha Diversity)是对单个样品中物种多样性的分析,包括Sobs指数、Chao指数、Ace指数、Shannon指数和Simpson指数等。本实验结果显示,各组细菌Alpha指数稀释曲线在测序条数达到15 000条以上时,趋于平台期,说明测序数据量合理,足够代表物种的丰富度。本研究结果显示,酵母壁多糖对断奶仔猪肠道菌群Alpha多样性影响不大,但在不考虑群落中每个物种的丰度前提下,0.30%酵母壁多糖组仔猪盲肠菌群最丰富;Beta多样性分析发现,当酵母壁多糖添加量为0.45%时,仔猪盲肠菌群与对照组差异最大,但0.15%组和0.30%组与对照组差异较小,且添加量0.15%的酵母壁多糖组相似性没其他组别好,这可能是由于胃肠道的复杂性,使得动物个体面对饲粮改变时,出现了个别不同的应答[11]。也可能是酵母壁多糖添加量较低,还没有达到改变肠道菌群的量,即只有当酵母壁多糖添加量达到一定程度时才能达到改变肠道菌群的效果。
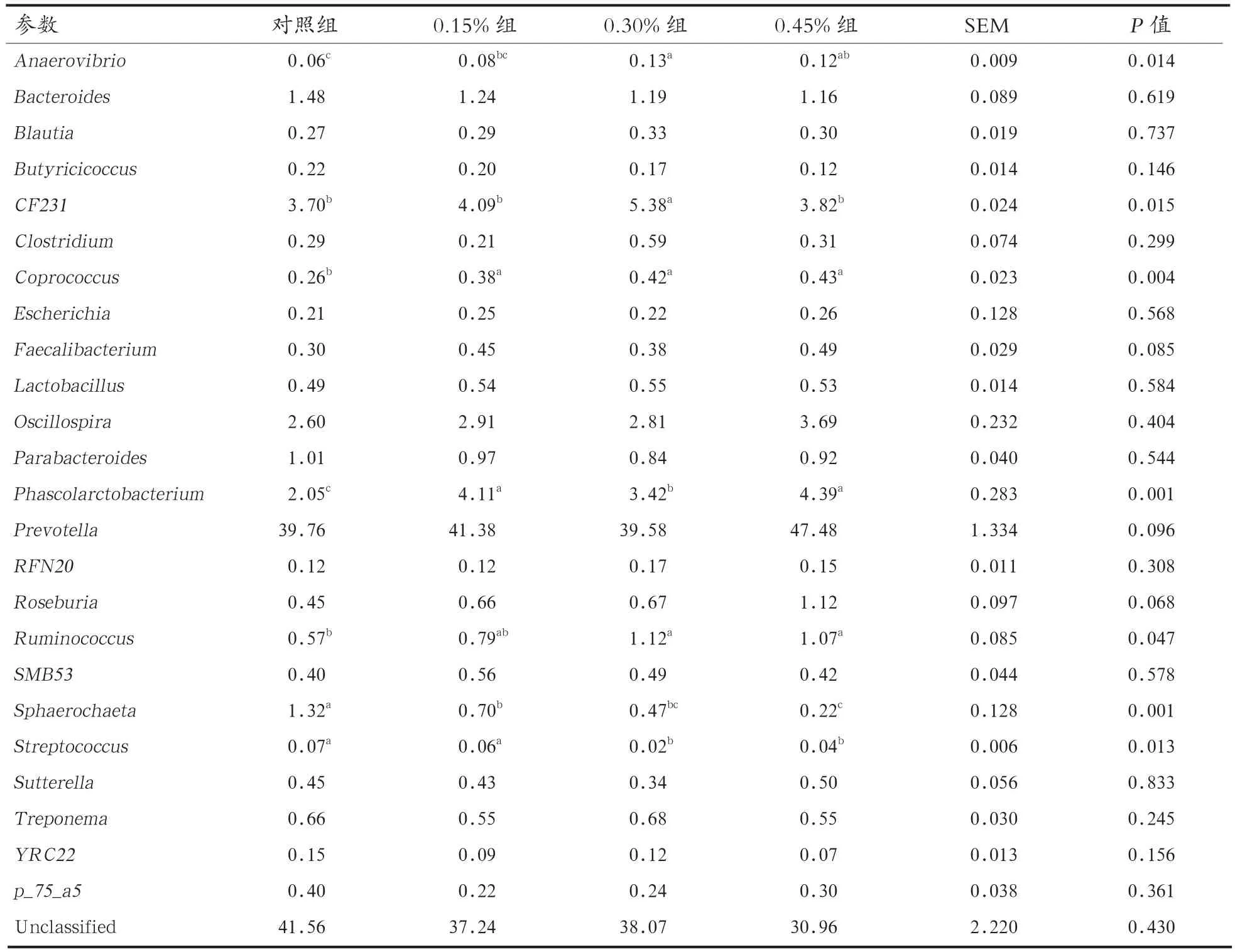
表4 不同水平酵母壁多糖对盲肠细菌组成的影响(属水平)
大量研究发现,肠道微生物主要以厌氧菌为主,严格厌氧菌的数量比兼性厌氧和需氧菌多2~3个数量级[12]。菌群结构门水平分析发现,肠道中优势菌门主要是拟杆菌门和厚壁菌门[13-17]。本研究发现,拟杆菌门、厚壁菌门和变形菌门为仔猪盲肠优势菌门,而软壁菌门、脱铁杆菌门、放线菌门和纤维杆菌门含量较低;相比于对照组,添加0.30%酵母壁多糖能显著降低断奶仔猪肠道拟杆菌门含量。这与Nakashimada等[18]研究结果相似。由于厚壁菌门和拟杆菌门具有降解多糖和促进机体能量吸收的作用[13]。本研究发现,0.30%和0.45%酵母壁多糖组盲肠厚壁菌门含量显著高于对照组。此外,添加0.45%酵母壁多糖还能显著提高断奶仔猪盲肠软壁菌门含量。进一步在属水平分析发现,普氏菌属为仔猪肠道主要菌属。普氏菌属的主要功能是降解粘蛋白和半纤维素、木聚素等植物性碳水化合物[19-20]。厌氧弧菌属和瘤胃杆菌属主要分解糖和纤维素[13],在反刍动物体内含量较高。本研究发现,添加0.30%和0.45%酵母壁多糖能显著提高仔猪盲肠厌氧弧菌属和瘤胃杆菌属含量。添加酵母壁多糖可能有利于仔猪盲肠对植物碳水化合物的消化利用。此外,酵母壁多糖还能显著提高断奶仔猪盲肠粪球菌属和考拉杆菌属的含量,并能显著降低链球菌属和Sphaerochaeta属含量。
4 结 论
当酵母壁多糖添加量达一定水平时,短期可改善断奶仔猪盲肠部分微生物含量。酵母壁多糖能显著提高断奶仔猪盲肠厚壁菌门含量以及瘤胃杆菌属和粪球菌属等优势菌群的含量。
[1] 李宁. 肠道菌群紊乱与粪菌移植[J]. 肠外与肠内营养, 2014, (4):193‐197.
[2] Gill S R, Pop M, DeBoy R T, et al. Metagenomic analysis of the human distal gut microbiome[J]. Science, 2006, 312(5778): 1355‐1359.
[3] Yatsunenko T, Rey F E, Manary M J, et al. Human gut microbiome viewed across age and geography[J]. Nature, 2012, 486(7402): 222‐223.
[4] 柳尧波, 凌泽春. 猪胃肠道微生物菌群的研究现状浅析[J]. 山东农业科学, 2011, (10): 90‐94.
[5] Duncan S, Scott K, Ramsay A, et al. Ef f ects of alternative dietary substrates on competition between human colonic bacteria in an anaerobic fermentor[J]. Appl Environ Microbiol, 2003, 69(2): 1136‐1142.
[6] Roberfroid M. Prebiotics: preferential substrates for specific germs?[J]. Am J Clin Nutr , 2001, 73(2): 406S‐409S.
[7] 贺琴, 王自蕊, 游金明, 等. 酵母壁多糖对断奶仔猪生长性能和小肠黏膜形态结构的影响[J]. 动物营养学报, 2016, (11): 3536‐3541.
[8] 贺琴, 王自蕊, 游金明, 等. 酵母壁多糖对断奶仔猪肠道挥发性脂肪酸和微生物菌群的影响[J]. 动物营养学报, 2017, (1): 177‐183.
[9] Yang L, Bian G, Su Y, et al. Comparison of faecal microbial community of lantang, bama, erhualian, meishan, xiaomeishan, duroc, landrace, and yorkshire sows[J]. Asian‐Austr J Anim Sci, 2014, 27(6):898‐906.
[10] 项云, 章啸君, 屠平光, 等. 仔猪肠道微生物宏基因组的研究分析[J]. 浙江畜牧兽医, 2016, (5): 1‐4.
[11] Ige B A. Probiotics use in intensive fi sh farming[J]. Afr J Microbiol Res, 2013, 7(22): 2701‐2711.
[12] Sommer F, Bäckhed F. The gut microbiota‐‐masters of host development and physiology[J]. Nat Rev Microbiol, 2013, 11(4): 227‐238.
[13] 姬玉娇, 祝倩, 耿梅梅, 等. 高、低营养水平饲粮对环江香猪结肠菌群结构及代谢物的影响[J]. 微生物学通报, 2016, (7): 1650‐1659.
[14] Kim H B, Borewicz K, White B A, et al. Longitudinal investigation of the age‐related bacterial diversity in the feces of commercial pigs[J]. Vet Microbiol, 2011, 153(1–2): 124‐133.
[15] Lu X, Lu P, Zhang H. Bacterial communities in manures of piglets and adult pigs bred with dif f erent feeds revealed by 16S rDNA 454 pyrosequencing[J]. Appl Microbiol Biotechnol, 2014, 98(6): 2657‐2665.
[16] Pajarillo E A B, Chae J P, Balolong M P, et al. Characterization of the fecal microbial communities of Duroc pigs using 16S rRNA gene pyrosequencing[J]. Asian‐Australas J Anim Sci, 2015, 28(4): 584‐591.
[17] Pajarillo E A, Chae J P, Balolong M P, et al. Pyrosequencing‐based analysis of fecal microbial communities in three purebred pig lines[J]. J Microbiol, 2014, 52(8): 646‐651.
[18] Nakashimada Y, Michinaka A, Watanabe K, et al. Brewer's yeast cell wall affects microbiota composition and decreases Bacteroides fragilis populations in an anaerobic gut intestinal model[J]. J Biosci Bioeng, 2011, 111(2): 178‐184.
[19] Mosenthin R. Physiology of small and large intestine of swine[J]. Asian‐Aust J Anim Sci, 1998, 11(5): 608‐619.
[20] Lamendella R, Domingo J, Ghosh S, et al. Comparative fecal metagenomics unveils unique functional capacity of the swine gut[J]. BMC Microbiol, 2011, 11(1): 1‐17.
Ef f ect of Yeast Cell Wall Polysaccharides on Cecum Bacteria Structure of Weaned Piglets by MiSeq Sequencing Technology
HE Qin1, WANG Zi‐rui1, YOU Jin‐ming1*, CHEN Li‐ling1,2, LU Ya‐fei1, LI Lan‐hai1
(1.Nutrition Feed Development Engineering Center of Jiangxi Province, College of Animal Science and Technology, Key Laboratory of Animal Nutrition in Jiangxi Province, Jiangxi Agricultural University, Jiangxi Nanchang 330045, China; 2.Jiangxi University of Traditional Chinese Medicine, Laboratory Animal Research Center for Science and Technology, Jiangxi Nanchang 330004, China)
An experiment was conducted to determine the effects of yeast cell wall polysaccharides on cecum bacteria structure of weaned piglets. A total of one hundred and eighty piglets with the similar genetic background, health condition, parity and body weight which weaned at 21 days of age were randomly allotted to 4 groups with 5 replicated each and 9 pigs in each replicate. Pigs in the four groups were fed control (without yeast cell wall polysaccharide), 0.15% of yeast cell wall polysaccharide, 0.30% yeast cell wall polysaccharide and 0.45% yeast cell wall polysaccharide, respectively. The experiment lasted for 21 days. After 21 days, the piglets were killed and the samples of cecum content were collected. Total bacterial DNA of 20 samples of pigs per group was extracted, and used to amplify PCR for 16Sr RNA gene V4 tag fragment and sequence using high‐throughput MiSeq technique. Based on the QIIME platform, the microbial community diversity in cecum content was compared. The results showed as follows: There were no significant differences on bacterial diversity in piglets receiving control diets and yeast cell wall polysaccharides. To a certain extent, dietary supplementation yeast cell wall polysaccharides can change the bacterial diversity, and without considering the abundance of each species, 0.30% yeast cell wall polysaccharide cecum flora was the most abundant. Bacteroidetes, Firmicutes, Spirochaetes and Proteobacteria were the dominant phyla in cecum content of piglets. Compared with the control group, dietary supplementation of 0.30% yeast cell wall polysaccharide signif i cantly decreased Bacteroidetes (P<0.05), 0.30% and 0.45% yeast cell wall polysaccharides significantly increased the amounts of Firmicutes. Added 0.45% yeast cell wall polysaccharide can signif i cantly increase the Tenericutes of cecum, in addition, Yeast cell wall polysaccharide could reduce the Proteobacteria of cecum, but there were no significant differences (P=0.066). Prevotella were the dominant genus in cecum content of piglets. The yeast cell wall polysaccharides enhance(P<0.05) Anaerovibrio, Ruminococcus, Coprococcus and Phascolarctobacterium, and reduced the Streptococcus of cecum(P<0.05). Accordingly, diets with yeast cell wall polysaccharides in short feeding‐period could improve the cecum bacteria structure of weaned piglets. Diets with yeast cell wall polysaccharides could enhance the Advantage bacterium of cecum, such as Firmicutes, Ruminococcus, Coprococcus,etc.
Yeast cell wall polysaccharides; Weaned piglets; Bacteria structure; Flora diversity
S828.5
A
10.19556/j.0258-7033.2017-09-125
2017-03-13;
2017-04-19
江西省重大科技专项(20143ACF60001);江西省生猪产业技术体系(JXARS-03-营养与饲料岗)
贺琴(1993-),女,江西莲花人,硕士研究生,研究方向为猪营养与饲料科学,E-mail:171836219@qq.com
*通讯作者:游金明,男,博士,教授,博士生导师,E-mail:youjinm@163.com
