利用Excel对粳稻全蛋白质组蛋白序列进行有序化管理
2017-09-15谢振华田继微
谢振华,田继微
(1.清华大学深圳研究生院健康科学和技术重点实验室,广东深圳518055;2.中南大学研究生院隆平分院,湖南长沙410083)
利用Excel对粳稻全蛋白质组蛋白序列进行有序化管理
谢振华1,田继微2
(1.清华大学深圳研究生院健康科学和技术重点实验室,广东深圳518055;2.中南大学研究生院隆平分院,湖南长沙410083)
利用Excel具有的数据管理功能实现粳稻全蛋白质组所有蛋白序列有序化管理,进而实现对粳稻全蛋白质组蛋白序列排序和归类。本研究将粳稻全蛋白质组48 905条蛋白序列的24个理化性质参数、蛋白名称、蛋白登录号码和蛋白序列组成数据矩阵,导入Excel中。根据蛋白不同理化性质参数进行排序,筛选得到特定理化性质的蛋白;根据蛋白名称排序,实现蛋白质家族成员和蛋白选择剪切变异体的系统归类排序;通过对粳稻全蛋白质组理化参数分布的可视化,促进对粳稻全蛋白质组更直观和全面的认识。
粳稻;全蛋白质组;蛋白;氨基酸;等电点;疏水性
谢振华,田继微.利用Excel对粳稻全蛋白质组蛋白序列进行有序化管理[J/OL].大麦与谷类科学,2017,34(4):10-15[2017-07-26]. http://kns.cnki.net/kcms/detail/32.1769.s.20170726.1859.005.html.
水稻是世界上栽种最广泛的谷类作物之一,其基因组相对较小,因此成为第一个完成全基因组测序的农作物,也是遗传分析、基因克隆及功能研究的理想模式植物[1-2]。基于水稻蛋白质组学研究数据,为科研人员提供有关作物对主要环境胁迫和病原反应有价值的信息,这可能会对提高作物产量和质量有所帮助,有利于解决全球粮食安全问题[3-4]。
一个生物全基因组上可能表达的所有蛋白质序列构成一个全蛋白质组[5-6]。随着大规模测序的发展,有1 171个真核生物的全蛋白质组信息已公布并供科研人员下载。全蛋白质组包涵了丰富的生物信息,通过对全蛋白质组分析,我们可以进行物种进化历史的研究[6-7]。然而,含有几千至几万条蛋白序列的全蛋白质组的处理对普通的生物学家而言是艰难的任务,导致目前对全蛋白质组的数据应用不多。
蛋白质疏水性、等电点(pI)、序列长度和分子量是独立于蛋白序列的信息,仅依赖于蛋白质的氨基酸组成。蛋白质的氨基酸组成和这些理化性质可以从氨基酸序列中计算得到,并被广泛应用于蛋白质的结构、功能、分类、蛋白-蛋白相互作用和蛋白质亚细胞定位等预测分析中[8-11]。以质谱(MS)为基础的鸟枪法是非常强大的蛋白质组分析方法,该方法在很大程度上依赖于全蛋白质组数据库[12-13];而常用的双向电泳方法也有一定的局限性,它很难分开非常酸性的、碱性的、小的、大的和疏水性蛋白[14]。通过相关方法的研究,进而实现对全蛋白质组的蛋白质序列理化性质的全面分析和展示,将促进蛋白质组学的发展。
本研究将粳稻全蛋白质组48 905条蛋白序列的理化性质参数作为坐标参数,将相应的蛋白登录号码作为坐标地址,并与相应的蛋白序列和蛋白名称输入数据矩阵同一行的不同单元格内,将每个蛋白名称、蛋白登录号码和蛋白序列与这些数据进行绑定,构成全蛋白质组的数据矩阵。全蛋白质组的数据矩阵导入Excel生成用于全蛋白质组管理的电子表格,利用Excel本身具有的数据管理功能实现全蛋白质组所有蛋白序列的有序化管理,进而实现对全蛋白质组中蛋白序列的排序和归类。
1 数据与方法
1.1 粳稻全蛋白质组Excel表格的建立
将粳稻全蛋白质组以FASTA格式从蛋白质资源库(universal protein resource,UniProt)下载[5]。用R语言从FASTA格式的粳稻全蛋白质组数据中提取出蛋白名称、蛋白登录号码和蛋白序列,然后通过去掉起始甲硫氨酸的方式生成蛋白MTS(Met-truncatedsequences)序列,并计算出所有MTS序列的氨基酸丰度和序列长度;用propas软件[15]和在线工具Compute pI/Mwtool(http://web.expasy.org/compute_pi/)计算出所有MTS序列的疏水性、等电点(pI)和分子量。最终将粳稻全蛋白质组48 905条蛋白序列的理化性质参数、蛋白名称、蛋白登录号码和蛋白序列组成数据矩阵,并导入Excel中,得到的电子表格称为“粳稻全蛋白质组数据表格”,并以附件形式在线存放(http://pan.baidu.com/s/1o8NrgJg)。
1.2 粳稻全蛋白质组有序化管理
通过对粳稻全蛋白质组Excel表格中数据进行排序,具体可分别以氨基酸丰度、序列长度、分子量、等电点(pI)和蛋白质的疏水性等参数数值大小进行排序,同时可完成以蛋白名称、蛋白登录号码和蛋白序列按字母顺序进行排序的粳稻全蛋白质组。
在对第1个理化性质参数进行排序之后,可以删除选定值以下或以上的数据行,然后对第2个理化性质参数进行排序,再删除选定值以下或以上的数据行,逐一完成不同参数的排序和数据排除,就可以从粳稻全蛋白质组中筛选出具备特定理化性质参数的全部蛋白质。
利用Excel表格的查找功能,对粳稻全蛋白质组实现检索和定位功能。可以用蛋白质的名称、部分名称、蛋白登录号码或一段蛋白序列进行检索,在粳稻全蛋白质组数据表格和重新排序的表格中定位要检索的蛋白。
1.3 粳稻全蛋白质组中蛋白理化性质分布的可视化
以粳稻全蛋白质组Excel表格中的数据为基础,用R语言可视化粳稻全蛋白质组的理化性质分布。在视图上,利用绿色的loess曲线和红色平均线来显示粳稻全蛋白质组理化性质的分布趋势。
2 结果与分析
2.1 粳稻全蛋白质组Excel表格的组织
粳稻全蛋白质组Excel表格有48 906行和28列,包含一个标题行和标题列,一个蛋白所有数据存放在同一行内,标题行上标题分别代表目标蛋白在全蛋白质组中的顺序号(NO.)、蛋白MTS序列各氨基酸 (Ala、Cys、Asp、Glu、Phe、Gly、His、Ile、Lys、Leu、Met、Asn、Pro、Gln、Arg、Ser、Thr、Val、Trp、Tyr)丰度、蛋白MTS序列的长度(SL)、蛋白MTS序列分子量(MW)、蛋白MTS序列等电点(pI)、蛋白MTS序列疏水性值(HP)、蛋白名称(Name)、蛋白注释(Annotation)和相应MTS序列(MTS)。
粳稻全蛋白质组中 Ala、Cys、Asp、Glu、Phe、Gly、His、Ile、Lys、Leu、Met、Asn、Pro、Gln、Arg、Ser、Thr、Val、Trp、Tyr的丰度,SL、MW、pI、HP的平均值分别为:0.101 6、0.021 3、0.049 9、0.056 0、0.033 6、0.080 1、0.026 9、0.039 3、0.042 5、0.091 4、0.019 3、0.029 8、0.063 0、0.032 4、0.077 1、0.083 0、0.047 8、0.068 2、0.014 2、0.022 5、305.2、33 470、7.87、-0.283。
2.2 粳稻全蛋白质组依据24个理化性质进行排序
通过对粳稻全蛋白质组Excel表格中Ala、Cys、Asp、Glu、Phe、Gly、His、Ile、Lys、Leu、Met、Asn、Pro、Gln、Arg、Ser、Thr、Val、Trp、Tyr的丰度,SL、MW、pI和HP所示的列分别排序,快速地实现对粳稻全蛋白质组48 905条蛋白MTS序列的各氨基酸丰度、序列的长度、分子量、等电点和疏水性值实现有序化管理。
通过对粳稻全蛋白质组有序化管理,发现5个粳稻蛋白的分子量和等电点是零值,这是因为它们的序列中含有不明确的氨基酸字符(x)所导致,所以这5个粳稻蛋白其他的理化性质参数也是不准确的。
通过粳稻全蛋白质组Excel表,以筛选富含赖氨酸(Lys)的蛋白为例,对粳稻全蛋白质组Excel表格中Lys列进行降序排序,快速系统地筛选出了粳稻中所有高赖氨酸蛋白,同时排序结果表明赖氨酸丰度≥18%的蛋白有95个,其中氨基酸长度>200的高赖氨酸蛋白有9个,分别是:Neurofilament triplet M protein-like protein(Q8LHS0)、Histone-like protein(Q851P9)、Os07g0184800 protein(Q8H4Z0)、Os05g0227600protein(Q84PC5)、Os05g0226900 protein (Q6AVC2)、Expressed protein (B7EFD0)、Expressed protein(Q6AST9)、Os01g0924900 protein(Fragment) (A0A0P0VC90)、Os03g0352300 protein (Fragment) (A0A0P0VY77)。其意义在于快速筛选到高赖氨酸蛋白,这种蛋白不仅具有营养和商业价值,也是建立高赖氨酸含量的转基因谷物的重要材料[16-17]。
小蛋白(≤100或200个氨基酸长度)广泛存在于古细菌、细菌、真核生物的全蛋白质组中,具有重要的生物学功能[18-19]。但至今未见系统地将全蛋白质组中所有小蛋白排列出来的方法。在本研究结果中,通过对粳稻全蛋白质组Excel表格中SL列进行升序排序,快速而系统地筛选出粳稻7 636个氨基酸长度≤100的小蛋白,21 650个氨基酸长度≤200的小蛋白。通过对粳稻全蛋白质组Excel表格中pI列进行排序,快速而系统地排列出粳稻4 547个pI≤5的酸性蛋白和8 872个pI≥10的碱性蛋白。这些结果为在粳稻蛋白质组学中研究设计更好的制备和分离条件提供了重要信息[20-22]。
2.3 粳稻全蛋白质组依据蛋白名称、登录号码和序列进行归类排序
通过对粳稻全蛋白质组Excel表格中Name、Annotation和MTS所示的列分别排序,快速地实现对粳稻全蛋白质组48 905条蛋白序列的蛋白名称、蛋白登录号码和蛋白序列按字母顺序进行的排序。
通过对蛋白名称的字母排序,可以对蛋白质家族成员和蛋白选择剪切变异体进行系统的归类排序,并迅速发现一些蛋白质家族成员和蛋白选择剪切变异体的序列长度、等电点和疏水性值的相关信息。对粳稻全蛋白质组蛋白质家族成员进行系统归类排序,可促进我们对粳稻全蛋白质组蛋白质家族的整体认识和家族成员的详细了解。
通过对蛋白氨基酸序列的排序,一些蛋白质家族成员或蛋白选择剪切变异体通常可以分组在一起,因为它们的N-末端氨基酸序列是相同的,但一些蛋白质家族成员或蛋白选择剪切变异体具有不同的N-末端氨基酸序列,它们分散地分布在对蛋白氨基酸序列排序后的表格中。
2.4 对粳稻全蛋白质组数据矩阵进行定位
利用Excel表格查找功能,可以用一段蛋白序列、蛋白名称或部分名称对粳稻全蛋白质组数据矩阵进行检索,检索的结果可以显示出目标蛋白在数据矩阵中的位置,从而实现定位功能。通过一段蛋白序列、蛋白名称或部分名称进行搜索定位,在按字母顺序排序的粳稻全蛋白质组Excel表格中,对任何蛋白家族或蛋白选择剪切变异体都能够快速识别和定位。
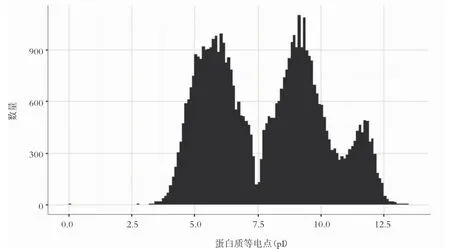
图1 粳稻全蛋白质组等电点的一维分布
2.5 粳稻全蛋白质组理化参数分布的可视化
为了更直观地显示粳稻全蛋白质组理化性质的分布,可对粳稻全蛋白质组的任何一个或两个理化参数的分布进行可视化。本研究仅展示等电点的一维分布(图1)、疏水性值的一维分布(图2)以及等电点-疏水性值二维分布(图3)。本文以Cys、Glu、Lys、Pro这4个氨基酸为例,均以蛋白序列长度-氨基酸丰度作二维分布图(图4),全部氨基酸的二维分布图组合成“序列长度-氨基酸丰度的二维分布图集”,以附件形式在线存放(http://pan.baidu. com/s/1o8NrgJg)。
全蛋白质组等电点的一维分布模式曾有报道[23-24]。本研究发现粳稻全蛋白质组的等电点的一维分布呈现3峰模式,与文献[24]中水稻蛋白质组等电点的一维分布模式有些差异,该文献水稻蛋白质组等电点的一维分布模式图中,在pI=8左右呈现一个小尖峰,在pI=11.3左右呈现一个小而平缓的宽峰,而本研究的图1中,在pI=8左右没有明显的峰形,在pI=11.0左右呈现一个中等的宽峰。这种差异源自下载的数据来自不同的数据库,文献[24]中水稻蛋白质组有83 159条蛋白序列。
本研究粳稻全蛋白质组的疏水性值的一维分布呈现单峰模式(图2)。粳稻全蛋白质组的等电点-疏水性值显示,在pI=11.0左右的碱性蛋白亲水性较好(图3)。
图4中4个蛋白序列长度(氨基酸的数目)-氨基酸丰度二维分布图尤如指纹图,全部20个氨基酸的蛋白序列长度-氨基酸丰度二维分布图组成粳稻全蛋白质组的指纹图谱。
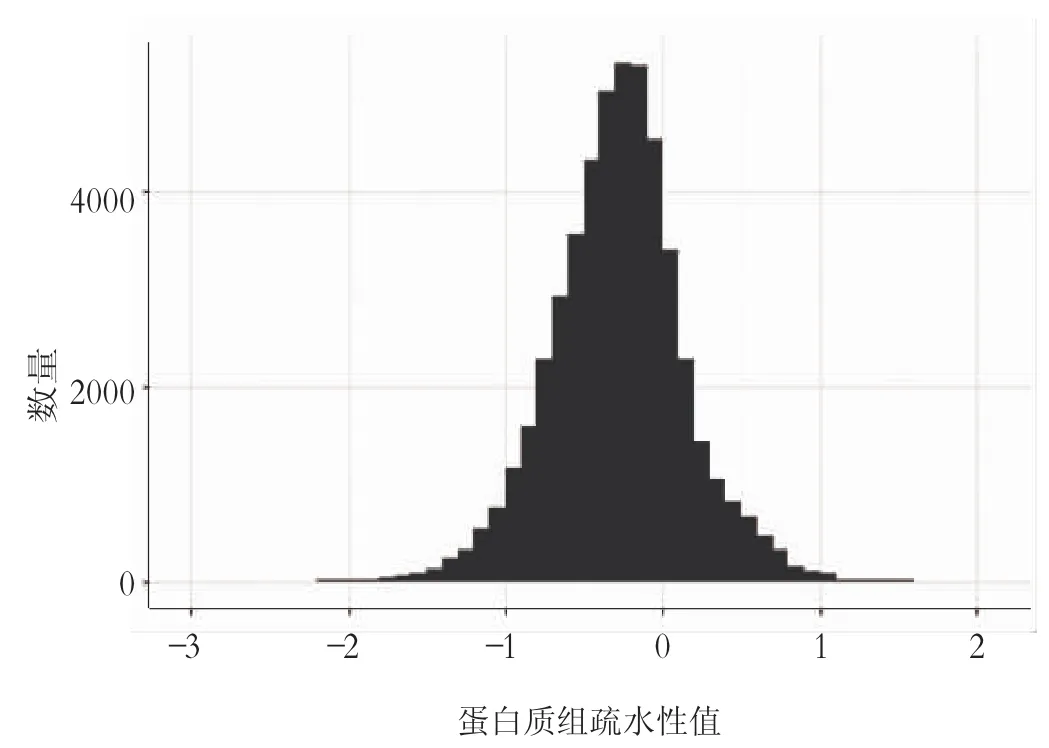
图2 粳稻全蛋白质组疏水性值的一维分布

图3 粳稻全蛋白质组等电点-疏水性值二维分布
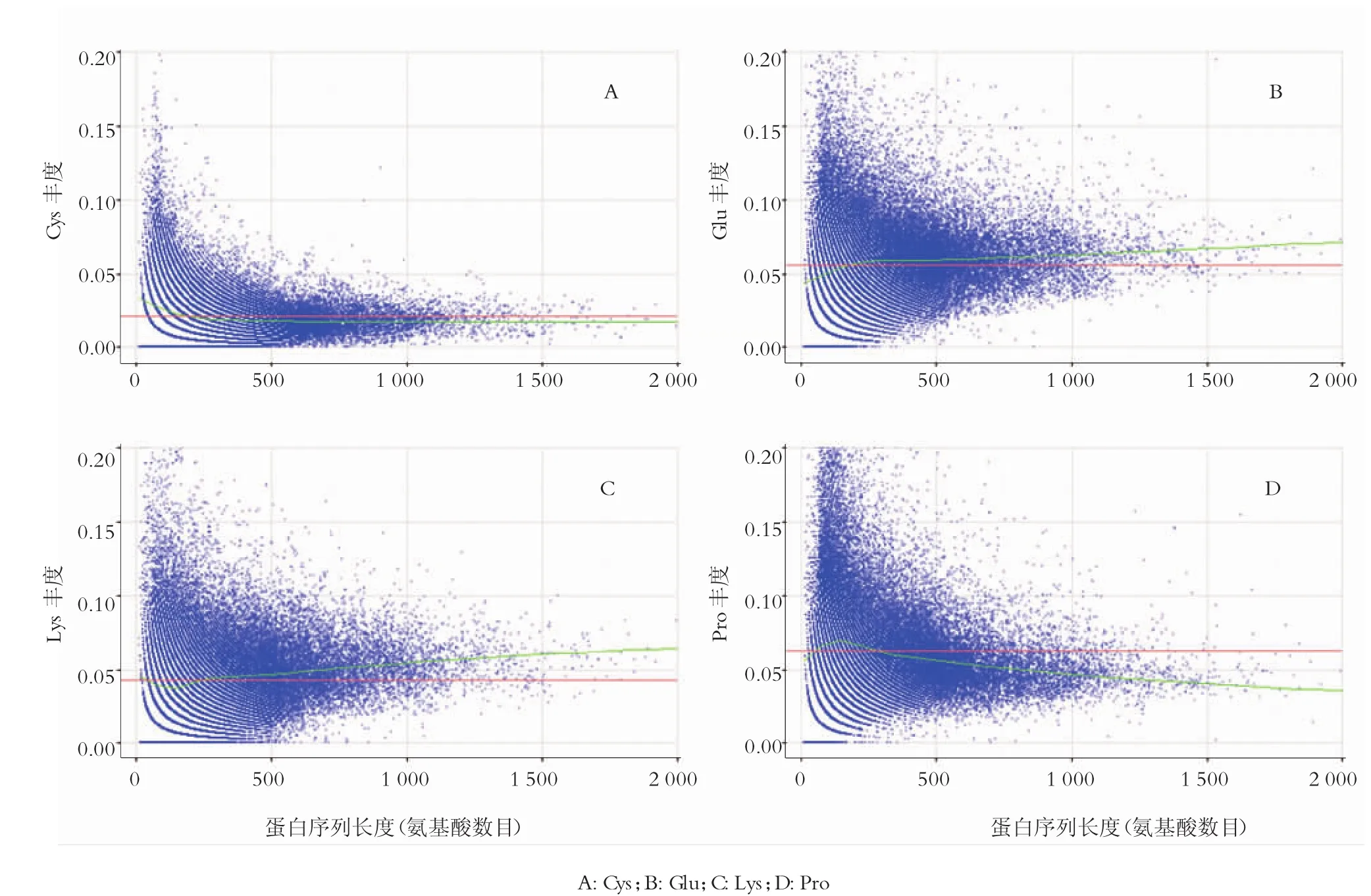
图4 粳稻全蛋白质组4个氨基酸的蛋白序列长度-氨基酸丰度二维分布
3 结论
网上生物数据库将蛋白质组中各个蛋白名称、蛋白登录号码和蛋白序列按一定格式组成一个个条目(entries),可以下载的一个物种全蛋白质组数据就是这个物种的所有蛋白条目的集合,由于条目总数达几千至几万,研究人员难以对其整体把握。本研究将粳稻全蛋白质组48 905条蛋白序列通过24个理化性质参数、蛋白名称、登录号码和序列进行系统的归类排序,使它们不但构成一个有机的整体,而且可以对这个整体进行操作。本方法可以让一个普通的生物学家对含有48 905条蛋白序列的粳稻全蛋白质组进行分析和处理,促进了研究人员对粳稻全蛋白质组更全面和深入的认识。这个粳稻全蛋白质组Excel表格还可以进行扩展,可把粳稻全蛋白质组所有蛋白相关的其他数字和文字信息整合到这个粳稻全蛋白质组Excel表格中,并对数字信息进行排序,也可对文字信息进行系统的归类排序。综上所述,采用Excel对粳稻全蛋白质组可以很好地进行有序化管理,该方法也可以应用到其他物种的全蛋白质组的管理与研究中。
[1]谢放鸣,彭少兵.杂交水稻在国外的发展历程与展望[J].科学通报,2016,61(35):3858-3868.
[2]SREENIVA N,BUTARDOV MJ R,MISRA G,et al.Designing climate-resilient rice with ideal grain quality suited for high-temperature stress[J].Journal of Experimental Botany, 2015,66(7):1737-1748.
[3]SINGH R,JWA N S.Understanding the responses of rice to environmental stress using proteomics[J].Journal of Proteome Research,2013,12(11):4652-4669.
[4]ZOUJ,LIUC,CHENX.Proteomics of rice in response to heat stress and advances in genetic engineering for heat tolerance in rice[J].Plant Cell Reports,2011,30(12):2155-2165.
[5]UP CONSORTIUM.Reorganizing the protein space at the U-niversal Protein Resource (UniProt)[J].Nucleic Acids Research,2012,40(Database issue):D71-75.
[6]MULDERNJ,KERSEY P,PRUESS M,et al.In silico characterization of proteins:UniProt,InterPro and Integr8[J]. Molecular Biotechnology,2008,38(2):165-177.
[7]CAFFREY B E,WILLIAMS T A,JIANG X,et al.Proteome-wide analysis of functional divergence in bacteria:exploring a host of ecological adaptations[J].PLoS One,2012,7(4): e35659.
[8]ROY S,MARTINEZD,PLATEROH,et al.Exploiting amino acid composition for predicting protein-protein interactions[J]. PLoS One,2009,4(11):e7813.
[9]GOODDM,MAMDOH A,BUDAMGUNTA H,et al.In silico proteome-wide aminoaCid and elemental composition(PACE) analysis of expression proteomics data provides a fingerprint of dominant metabolic processes[J].Genomics,Proteomics& Bioinformatics,2013,11(4):219-229.
[10]HUANG CH,CHOUS Y,NG K L.Improving protein complex classification accuracy using amino acid composition profile[J].Computers in Biology and Medicine,2013,43(9): 1196-1204.
[11]HAYATM,KHANA.WRF-TMH:predicting transmembrane helix by fusing composition index and physicochemical properties ofaminoacids[J].AminoAcids 2013,44(5):1317-1328.
[12]ALHAIDERA A,BAYOUMY N,ARGOE,et al.Survey of the camel urinary proteome by shotgun proteomics using a multiple database search strategy[J].Proteomics,2012,12(22): 3403-3406.
[13]MARTINS-de-SOUZA D,GUEST P C,GUEST F L,et al. Characterization of the human primary visual cortex and cerebellum proteomes using shotgun mass spectrometry-data-independent analyses[J].Proteomics,2012,12(3):500-504.
[14]RABILLOUDT,CHEVALLETM,LUCHE S,et al.Two-dimensional gel electrophoresis in proteomics:Past,present and future[J].Journal of Proteomics,2010,73(11):2064-2077.
[15]WUS,ZHUY.ProPAS:standalone software to analyze protein properties[J].Bioinformation,2012,8(3):167-169.
[16]孙晓波,房 瑞,余桂红,等.辣椒高赖氨酸蛋白基因Cflr全长cDNA的克隆及其组织表达特征 [J].园艺学报, 2008,35(9):1310-1316.
[17]YUE J,LI C,ZHAOQ,et al.Seed-specific expression of a lysine-rich protein gene,GhLRP,fromcotton significantly increases the lysine content in maize seeds[J].International Journal of Molecular Sciences,2014,15(4):5350-5365.
[18]SUM,LING Y,YUJ,et al.Small proteins:untapped area of potential biological importance[J].Frontiers in Genetics,2013 (4):286.
[19]YANGX,TSCHAPLINSKI TJ,HURSTGB,et al.Discovery and annotation of small proteins using genomics,proteomics, and computational approaches[J].Genome Research,2011,21 (4):634-641.
[20]URQUHART B L,CORDWELL S J,HUMPHERY-SMITH I.Comparison of predicted and observed properties of proteins encoded in the genome of Mycobacteriumtuberculosis H37Rv [J].Biochemical and Biophysical Research Communications, 1998,253(1):70-79.
[21]WUWW,WANGG,YUMJ,et al.Identification and quantification of basic and acidic proteins using solution-based two-dimensionalprotein fractionation and label-freeor 18O-labeling mass spectrometry[J].Journal of Proteome Research,2007,6(7):2447-2459.
[22]ADHIK A S,MANTHENA P V,SAJWANK,et al.A unified method for purification of basic proteins[J].Analytical Biochemistry,2010,400(2):203-206.
[23]WEILLER G F,CARAUX G,SYLVSETER N.The modal distribution of protein isoelectric points reflects amino acid properties rather than sequence evolution [J].Proteomics, 2004,4(4):943-949.
[24]WUS,WANP,LI J,et al.Multi-modality of pI distribution in whole proteome[J].Proteomics,2006,6(2):449-455.
·简讯与信息·
本刊加入有关数据库的特别声明
为了适应我国信息化建设的需要和扩大作者学术交流渠道,实现期刊编辑、出版工作的网络化与数字化,提高作者所发表论文的被引频次与影响力,本刊已加入《中国学术期刊(光盘)》、“中国期刊网”、“万方数据-数字化期刊群”、“重庆维普”:“中文期刊数据库”与超星期刊“域出版”平台。作者无需支付网络编审费。作者著作权使用费与本刊稿酬由本刊编辑部一次性给付作者。如有作者不同意将文章编入上述数据库,请在来稿时声明,本刊将作适当处理。所有刊载文献以各种形式转载时请注明来源于本刊。
《大麦与谷类科学》杂志编辑部
Using an Excel Table to Orderly Manage All 48905 Proteins in the Complete Proteome of Japonica Rice
XIE Zhen-hua1,TIAN Ji-Wei2
(1.The Shenzhen KeyLaboratoryofHealth Sciencesand Technology,Graduate SchoolatShenzhen,Tsinghua University,Shenzhen 518055,China;2.LongpingBranch ofGraduate School,CentralSouth University,Changsha,410083,China)
Owing to its function of data management,Excel can be used to sort and cluster all protein sequences of the complete proteome of japonica rice.In this study,a data matrix was constructed,comprising 24 physicochemical parameters,names,accession numbers,and sequences of 48 905 proteins in the complete proteome of japonica rice;and this data matrix has been imported into an Excel table for orderly management,clustering,and querying.Any proteins with some particular physicochemical features can be screened out from the complete proteome of japonica rice by orderly management;all members of a protein family or protein splice variants can be systematically clustered by alphabetically sorting the name column.Such an Excel table provides an overview of the complete proteome of japonica rice by visualizing the distribution of the physicochemical parameters of all proteins.Therefore,this study creates a tool that is instrumental in comprehensive and in-depth understanding of the complete proteome of Japonica rice.
Japonica rice;Complete proteome;Protein;Amino acid;Isoelectric point;Hydrophobicity
Q51
A
1673-6486-20170333
2017-03-07
深圳市科技创新委员会资助项目(JCYJ201404171158402 67和JCYJ20150518162154828)。
谢振华(1964—),男,博士,讲师,主要从事细胞分子生物学研究。E-mail:xiezh@sz.tsinghua.edu.cn。
