GC联合GC-MS测定果蔬汁中11种有机磷农药的残留量
2017-09-12黄佳新
黄佳新
GC联合GC-MS测定果蔬汁中11种有机磷农药的残留量
黄佳新
(中国检验认证集团 广东有限公司番禺分公司,广东 广州 511400)
建立了气相色谱仪(GC)与气相色谱质谱联用仪(GC-MS)结合测定果蔬汁中11种有机磷农药残留量的分析方法。优化了样品前处理方法,同时优化了GC与GC-MS分析条件。样品以乙酸乙酯提取,添加氯化钠与无水硫酸镁优化萃取条件,以GCB/PSA小柱净化后供GC和GC-MS分析。该方法简便、快速、准确,11种有机磷农药在0.05~0.4 mg/L范围内,显示了良好的线性关系,相关系数均大于0.995,平均加标回收为81.29%~111.74%,回收实验的相对标准偏差(RSD,=6)在1.9%~8.6%之间。
果蔬汁;有机磷农药;GC-MS;GC
随着人们生活水平的提高,人们对日常饮品的营养安全健康愈发关注和重视。果蔬汁在口感和营养方面都接近新鲜果蔬,且食用更为方便,并且有一定的保健价值,受到各年龄阶段人们的喜爱。然而这种以果蔬为原料的产品的安全性势必受到果蔬原料质量优劣的威胁。
有机磷农药在我国农业生产过程中使用量十分广泛,因此果蔬中不可避地会有该类农药残留[1]。使用农药残留超标的食品,虽然不一定会导致急性中毒,但很可能会导致人体慢性中毒,引起人体器官病变,诱发癌症[2-4]。
目前,有机磷农药残留的检测方法有气相色谱法(GC)[5]、气相色谱-质谱法(GC-MS)[6,7]、超临界流体色谱法[8]、气相色谱串联质谱法(GC-MS/MS)、液相色谱-串联质谱法(LC-MS/MS)[9]等等。其中GC-FPD法具有灵敏度高,杂峰干扰少,成本低廉的优点,但由于只依靠保留时间定性,对某些无法分离开的有机磷分辨率较低;而GC-MS法则有高分辨率,高准确性的优点,但成本相对较高,针对个别有机磷响应较低,难以准确定量的缺点。
本实验方法依照实验室现有条件,将GC-FPD法与GC-MS法结合应用,同时优化前处理方法,建立以GC-FPD法初筛及最后定量,阳性样以GC-MS法确证定性的方法。该法具有快速准确,灵敏度和精密度高,重现性好,实用经济等优点,可为食品中有机磷农药残留的检测与监测提供参考与支持。
1 试验部分
1.1 仪器试剂
气相色谱仪(配有FPD火焰光度检测器)(Agilent 7890A,美国安捷伦公司),气相色谱质谱联用仪(Agilent7890A-5975C,美国安捷伦公司),大容量冷冻离心机(Eppendorf 5810r,德国艾本德公司),旋蒸蒸发仪(Heidolph ML/G3B,德国海道夫公司),氮吹仪(I WILL T&D MTN-2008W,天津艾维欧公司);
果蔬汁:市售;
11种有机磷农药标准品:敌敌畏,速灭磷,久效磷,甲拌磷,乐果,甲基对硫磷,杀螟松,马拉硫磷,倍硫磷,对硫磷,克线磷(均为1ml浓度1g/L丙酮溶液,购自农业部环境保护科研监测所)
乙酸乙酯,乙腈,正己烷,丙酮为色谱纯(美国默克公司);无水硫酸镁、无水硫酸钠、氯化钠为分析纯(广州化学试剂厂)。
1.2 标准溶液配制
分别吸取100 μL 1 g/L标准溶液至10 mL容量瓶中,用乙酸乙酯定容,配成10 mg/L单一标准溶液,再按需吸取100 μL 1g/L标准溶液至同一10 ml容量瓶中,定容后配置成所需10 mg/L混合标准溶液,均保存至4°C环境中。测试时,将单一标准溶液及混合标准溶液按需求稀释成单一及混合标准工作液。
1.3 样品前处理
量取已混匀的果蔬汁样品5 g于50 mL离心管中,加入15 mL乙酸乙酯,添加2 g氯化钠及1 g无水硫酸镁,涡旋混匀,超声提取10 min,离心5 min,取上层有机相于100 mL旋蒸瓶中。再加入15 mL乙酸乙酯重复以上步骤提取一次,合并两次提取液,旋蒸至2~3 mL。将浓缩液加入已用5 mL乙酸乙酯活化的GBC/PSA固相萃取小柱,再用6 mL乙酸乙酯洗脱小柱,收集所有浓缩液及洗脱液,35℃以下氮吹浓缩至近干,定容至1 mL上机分析。
1.4 仪器条件
1.4.1 气相色谱(FPD检测器)条件
色谱柱:HP-5MS毛细管色谱柱(30 m×0.32 mm×0.25μm);载气为高纯氮气,流速为2.0 mL/min,进样口温度260 ℃,进样方式为不分流进样,到分流口的吹扫流量为25 mL/min。色谱柱升温程序:60 ℃保持2 min, 20 ℃/min程序升温至120 ℃, 10℃/min程序升温至180 ℃,再以2 ℃/min程序升温至190 ℃,再以15 ℃/min程序升温至升至300度,保持10 min。检测器温度245 ℃,氢气流量75 mL/min,空气流量100 mL/min,尾吹气流量60 mL/min。
1.4.2 气相色谱质谱联用条件
色谱柱:HP-5MS毛细管色谱柱(30 m×0.32 mm×0.25μm);载气为高纯氦气,流速为1.0 mL/min,进样口温度260 ℃,进样方式为不分流进样,到分流口的吹扫流量为25 mL/min。色谱柱升温程序:60℃保持2 min, 20 ℃/min程序升温至120 ℃, 10℃/min程序升温至180 ℃,再以2 ℃/min程序升温至195 ℃,再以5℃/min程序升温至205 ℃,再以15 ℃/min程序升温至升至300 ℃,保持10 min。质谱条件:电子轰击离子源;电子能量70 eV;GC-MS接口温度280 ℃;离子源温度230 ℃;MS四级杆温度150 ℃。
2 结果与讨论
2.1 前处理方法的优化
依据“相似相溶”原则选取实验室常用的提取试剂,包括丙酮、乙酸乙酯、丙酮-正己烷(/, 1∶1)、乙腈等为待选试剂。丙酮为经典的提取试剂选择,石油醚与丙酮均为经典的提取试剂,提取效果好,但针对性不强,提取过程会带出许多杂质,对后阶段的净化造成一定的困难。丙酮-正己烷(/, 1∶1)对极性较强的农药回收率较低[10],如敌敌畏、甲基对硫磷等。乙腈与乙酸乙酯极性均比丙酮弱,又比正己烷强,提取过程可避免许多杂质干扰,选择性较强。考虑到果蔬汁为液体样品,乙腈与水互溶,会导致大量水的引入提取液,且相对于乙酸乙酯来说,乙腈对人体毒性更强,故而参照可乐中有机磷农药测定国标方法[11],选用乙酸乙酯作为提取试剂。经实验表明,提取液中加入氯化钠后能明显改善分层效果,不加入氯化钠会难以分离有机相。相比于无水硫酸钠,加入适量无水硫酸镁不会轻易造成结块,而且加入无水硫酸镁涡旋混匀后会轻微放热,可提高提取效率。故而在加入提取试剂后添加2 g氯化钠及1 g无水硫酸镁。
PSA固相萃取小柱具有弱阴离子交换、极性螯合作用,可清除极性基质成分,而GCB石墨碳黑小柱能有效吸附色素,经对比实验表明,相比单独使用活性炭小柱或者使用Florisil/GCB小柱,PSA/GCB小柱在保证回收率的同时有更好的净化效果,基线响应更低。样品净化情况谱图比较如图1所示,基线最低的为使用Florisil/GCB小柱所得的图谱。
由于所选测试的有机磷农药有易挥发的药物敌敌畏,在旋蒸浓缩时不可蒸干,实验过程中发现,旋蒸操作中蒸干溶液,会导致敌敌畏回收率大幅度降低。氮吹过程也需要控制好气流强度和水浴温度,不可吹干,温度控制在35 ℃以下能保证较好的回收率。
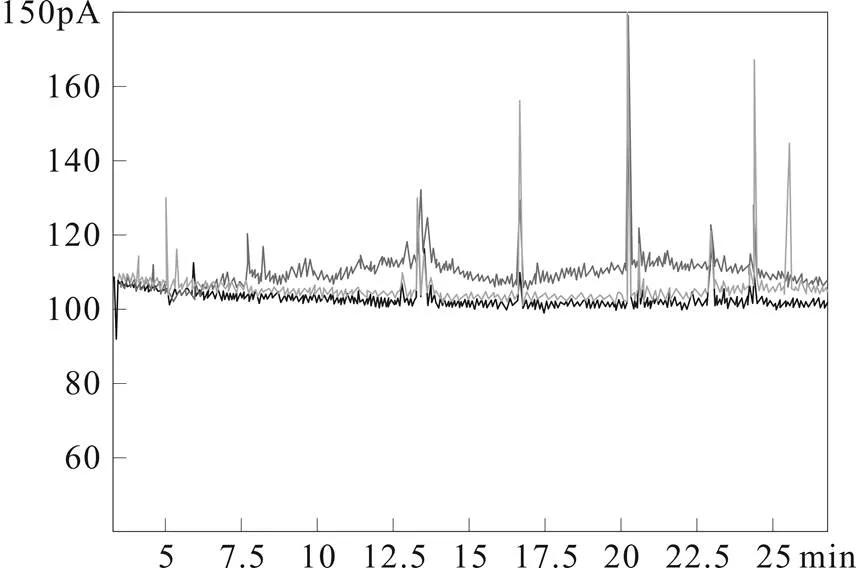
图1 三种固相萃取柱净化效果

图2 sim模式下11种有机磷农药(0.1 mg/L)的总离子流色谱图
2.2 色谱质谱条件的优化
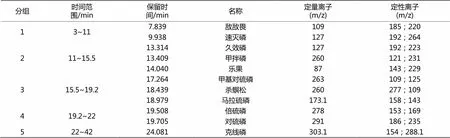
表1 11种有机磷农药的分组和特征离子
试验采用气相色谱质谱联用仪选择离子扫描模式进行分析。先将11种有机磷农残混标及单标分别进行全扫描分析,确定其保留时间及出峰顺序,同时对每一种农残进行单独分析,分别选择丰度大、特异性强的离子作为定量离子及定性离子。然后在SIM模式下,根据目标组分的保留时间进行分组扫描,以减少试样中物质干扰,提高灵敏度。表1列出了11种有机磷农药的分组和特征离子以及分离效果。图2为SIM模式下0.1 mg/L 11种有机磷农药的选择离子监测总离子流色谱图。在气相色谱中无法分离的几种有机磷在GC-MS图谱中仍有相近的保留时间,但是在SIM模式下,由于选择的定量离子不同,它们仍能被有效定性。
试验采用气相色谱仪(FPD)进行分析。将11种有机磷农残混标及单标分别进样,确定其保留时间及出峰顺序。浓度为0.1 mg/L的11种有机磷混合标准溶液气相色谱(FPD)色谱图如图3所示。
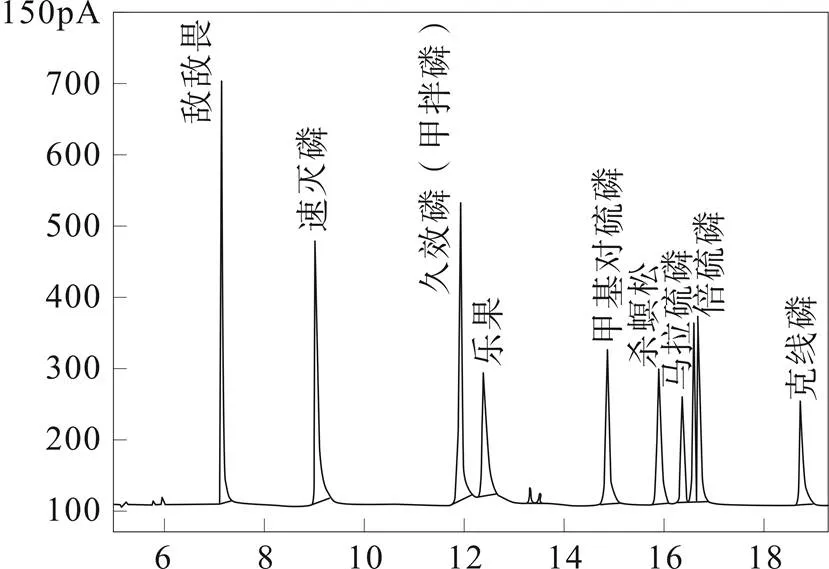
图3 11种有机磷标准溶液(0.1 mg/L)气相色谱图
可以看到,11种有机磷在此条件下都有良好的响应,相比于气质联用法,11种有机磷低浓度时在气相法有更干净的背景,更高的信噪比,个别有机磷的信噪比更是大幅度提高。但是甲拌磷与久效磷、倍硫磷磷与对硫磷无法很好的分离。尝试通过优化流速、升温程序等等仪器条件仍无明显改善。将11种有机磷农残分成两组进行单独定量,甲拌磷与倍硫磷为一组,其余九种分为一组,可保证色谱峰的分离,保证定量结果的准确性,但无法定性,需要采用气质联用方法进行阳性确证。所以本方法中气相色谱法用于筛选及定量,阳性样品采用气质方法进行确证,可保证实验速度与实验结果。
2.3 标准曲线、相关系数、回收率及精密度
取甲拌磷、倍硫磷混合标准溶液及其余九种有机磷混合标准溶液分别稀释成质量分数为25、50、100、200和400μg/kg的系列标准工作溶液,并上GC进行测定。以各农药的浓度为横坐标,峰面积响应为纵坐标,分别绘制标准曲线;选取空白的果蔬汁样品,进行了加标量为0.05、0.1、0.2μg的加标回收实验,每个加标水平进行平行测定6次。11种有机磷农药的线性方程、相关系数、回收率及精密度如表2所示。
由表中可见,各化合物均有良好的线性关系,相关系数均大于0.995;11种有机磷农药的平均回收率在81.29%~111.74%之间,相对标准偏差(RSD,=6)在1.9~8.6之间,符合相关标准规定。

表2 11种有机磷农药的线性方程、相关系数、回收率及精密度
3 结论
本实验建立了采用乙酸乙酯提取,GCB/PSA小柱净化,结合GC及GC/MS,通过优化前处理方法及仪器条件,建立针对所选11种有机磷农药的准确定性及定量方法。本方法采取先由GC初筛,再由GC-MS进一步定性,最后气相色谱定量的方式,使有机磷定性定量更为准确。该方法简便经济,省时省力,定性定量更为准确可靠,灵敏度高,适用于多有机磷农药的同时检测,为有机磷农药的检测提供一种有效的定性定量方法。
[1] 吴烨,周伦敏,陆煜养,等.农药降解对蔬菜中有机磷农药残留检测结果的影响[J].中国卫生检验杂志,2014,24(19): 2846.
[2] QIN G F,LI Y B,CHEN Y, et al.Pesticide residues determination in China vegetables in 2010—2013 applying gas chromatography with mass spectrometry[J]. Food research international,2015,72: 161.
[3] 马莉莉,张承聪,吴晓波,等.固相萃取-高效液相色谱法同时测定果蔬中9种农药残留[J].理化检验:化学分册,2010,46(7):825-828.
[4] 祖正贤.人参中农药多残留检测方法的建立及膳食风险评估研究[D].长春:吉林农业大学,2014:12-18.
[5] ANDREIA N O J,DENISE C M,FEMANDA C S G. Pesticide residues in cashew apple, guava,kaki and peach: GC-IECD,GCFPD and LC-MS /MS multiresidue method validation, analysis and cumulative acute risk assessment[J]. Food chemistry,2014,30( 5) : 195.
[6] 王飞,蒋闳,郑屏.食品中有害物质残留分析检测技术进展[J].安徽化工,2007,33(6):3-7.
[7] 袁鹏辉,陈明晓,董军,等.固相萃取-气相色谱/质谱选择离子检测法测定水中27 种有机农药[J].分析试验室,2017,36(3):322-328.
[8] NAM K,KING J W.Coupled SFE/SFC/GC for the trace analysis of pesticide residues in fatty food samples[J]. Journal of high resolution chromatography,1994,17( 8) : 577.
[9] 周荣,曹赵云,赵肖华,等.分散固相萃取- 分散液液微萃取/气相色谱- 串联质谱法测定蔬菜中19 种有机磷农药残留[J].分析测试学报,2017,36(1):67-72.
[10]刘开,孔祥虹,何强,等.GC-GC/MS法测定植物提取物种39种有机磷类农药的残留量[J].分析实验室,2015,34(12):1432-1437.
[11]中国人民共和国国家卫生和计划生育委员会,中华人民共和国农业部,国家食品药品监督管理总局 GB 23200.40-2016《食品安全国家标准 可乐饮料中有机磷、有机氯农药残留量的测定 气相色谱法》[S].2017-06-18.
Determination of 11 Kinds of Organophosphorus Pesticides Residues in Fruit and Vegetable Juice by GC and GC-MS
(China Certification & inspection group Guangdong Co., Ltd. Panyu branch, Guangdong Guangzhou 511400, China)
An analysis method was established for the determination of 11 kinds of organophosphorus pesticides residues in fruit and vegetable juice by using gas chromatograph(GC) and gas chromatograh-mass spectrometry(GC-MS). The GC and GC-MS analysis conditions and sample preparation methods were optimized. The samples were first extracted with acetate, sodium chloride and anhydrous magnesium sulfate were added to optimize the conditions of extraction, then they were purified by GCB/PSA column and detected by GC and GC-MS. The method is simple, fast and accurate.The results show that there is good linearity in the range of 0.05~0.4 mg/L,the correlation coefficients are higher than 0.995,the recoveries are in the range of 81.29% to 111.74% with the relative standard deviations (RSD,=6) of the recoveries between 1.9% and 8.6%..
Fruit and vegetable juice;Organophosphorus pesticides; GC-MS; GC
O 657
A
1671-0460(2017)08-1726-04
2017-06-14
黄佳新(1987-),男,广东省广州市人,实验员,2010年毕业于华南农业大学食品学院,研究方向:从事进出口食品安全检测。E-mail:157131700@qq.com。
