不同年龄组儿童噬血细胞综合征的临床特点及预后研究
2017-09-12贺钰磊
李 蕙,李 丹,贺钰磊,史 宏,周 敏
(成都市妇女儿童中心医院血液肿瘤科 610091)
论著·临床研究
不同年龄组儿童噬血细胞综合征的临床特点及预后研究
李 蕙,李 丹,贺钰磊,史 宏,周 敏△
(成都市妇女儿童中心医院血液肿瘤科 610091)
目的 探讨不同年龄组儿童噬血细胞综合征(HPS)的临床特点与预后因素分析。方法 将85例HPS患儿按年龄分为0~2岁(A组)、>2~8岁(B组)和>8~14岁(C组),从病因、病理、临床症状、实验室检查及预后等方面进行回顾性分析。结果 A、B和C组病因均以EB病毒感染为主,感染阳性率C组明显高于A、B组;骨髓检查粒细胞红细胞比例(粒红比)小于1.0的发生率,A组高于C组;颈部淋巴结肿大发生在B组中多见,肝脾肿大在C组中最少见;浆膜腔积液发生在B组多见;实验室检查乳酸脱氢酶(LDH)B组高于A组,以上差异均有统计学意义(P<0.05)。其他临床症状和实验室检查结果在组之间差异无统计学意义(P>0.05)。存活与死亡患儿比较,浆膜腔积液、三酰甘油(TG)、CD4/CD8及骨髓检查粒红比比较,差异有统计学意义(P<0.05),将上述因素进行Logistic分析,显示TG>3.5 mmol/L、CD4/CD8<1.24、骨髓粒红比小于1.02是影响儿童HPS预后的危险因素(P<0.05)。结论 儿童HPS有部分临床特点与发病年龄有关,TG、CD4/CD8及骨髓粒红比水平是影响预后的危险因素。
淋巴组织细胞增多症;噬血细胞性;年龄因素;临床特点;预后;危险因素
噬血细胞综合征(hemophagocytic syndrome,HPS)又称噬血细胞性淋巴组织细胞增生症(hemophagocytic lymphohistiocytosis,HLH),是以发热、肝脾肿大、全血细胞减少、凝血障碍,以及骨髓或其他组织中可发现噬血细胞为特征的一类综合征[1]。该病分为原发性和继发性,后者又分为感染相关性、肿瘤相关性及自身免疫性疾病等所致HPS[2]。儿童HPS病情常起病急、进展迅速、预后差,容易误诊。为提高对该病的认识及诊疗水平,本研究将本院2003年6月至2016年12月收治的85例患儿,进行临床特点及预后分析,现将研究结果报道如下。
1 资料与方法
1.1 一般资料 收集本院儿童血液科2003年6月至2016年12月确诊的HPS患儿85例,纳入标准参照国际组织细胞学会制订的HLH-94(2004年以前)及HLH-2004方案(2004年以后)诊断标准(符合以下标准中的1条则诊断成立)[3-5]。(1)分子生物学符合HLH。(2)符合初诊标准及新诊断标准中的5项。①初诊标准:发热,脾肿大,全血细胞减少,累及大于或等于2个细胞系,血红蛋白小于90 g/L,血小板小于100×109/L,中性粒细胞小于1.0×109/L;高三酰甘油(TG)血症和(或)低纤维蛋白原血症,TG≥3.0 mmol/L,纤维蛋白原小于或等于1.5 g/L;骨髓或脾或淋巴结活检发现噬血细胞存在(增多),无恶性疾病证据。②新诊断标准:自然杀伤细胞(NK)细胞活性减低或缺乏;铁蛋白大于或等于500 μg/L;可溶CD25或者可溶白细胞介素-2(IL-2)受体大于或等于2 400 U/mL。按年龄将85例患儿分为0~2岁(A组,42例,男20例,女22例),>2~8岁(B组,32例,男15例,女17例),>8~14岁(C组,11例,男4例,女7例)。所有患儿监护人均知情同意,并经本院伦理委员会审核通过。3组患儿性别差异无统计学意义(P>0.05)。
1.2 疗效标准 参照国际组织协会2004年提出的疗效标准[3,6-7]。(1)临床有效(在初始诱导阶段符合以下标准时判定临床治疗有效):体温正常;脾脏缩小;血小板大于或等于100×109/L;纤维蛋白原正常;血清铁蛋白下降超过25%。(2)临床缓解(在诱导治疗结束后如达以下标准,判定为临床缓解):体温正常;脾脏大小恢复正常;血细胞计数恢复正常(血红蛋白大于或等于90 g/L,血小板大于或等于100×109/L,中性粒细胞绝对值大于或等于0.5×109/L);血TG恢复正常(<3 mmol/L);血清铁蛋白正常(<500 μg/L);脑脊液检查正常(治疗前脑脊液检查异常者);血浆可溶性 CD25下降。(3)疾病活动(如患者未达到临床缓解标准时,则认为疾病活动)。(4)疾病再活动(如患者达临床缓解后再次表现下面中的3条及3条以上时则判定为疾病再次活动):发热;脾大;血小板小于100×109/L;血TG升高(≥3 mmol/L);纤维蛋白原下降(<1.5 g/L);噬血细胞现象;铁蛋白升高;血浆可溶性CD25升高。如患者出现新的中枢神经系统症状,单独即可诊断疾病再活动。
1.3 基因检查 检查标本送至北京海斯特临床检验所,采用PCR和二代测序的方法进行嗜血相关基因检查。检测标本中PRF1、UNC13D、STX11、STXBP2、XIAP、SH2D1A、Rab27a、AP3B1、LYST、ITK这10个基因的编码区全长及附近转录剪接位点的突变分析。
1.4 治疗方案 2004年前多采用肾上腺皮质激素(地塞米松或甲泼尼龙)+大剂量丙种球蛋白400 mg·kg-1·d-1,3~5 d。2004年后开始HLH2004方案[包括地塞米松+环孢素A+依托泊苷(VP16)]+大剂量丙种球蛋白免疫治疗。对明确有病毒感染的患儿予更昔洛韦、干扰素等抗病毒治疗,对明确有细菌感染的患儿予抗菌药物抗感染治疗。肝功异常者予保肝治疗,贫血、血小板减少、凝血功能异常者予输注红细胞悬液、血小板、纤维蛋白原、新鲜冰冻血浆。
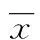
2 结 果
2.1 发病年龄与性别 本研究收集患儿共85例,其中A组42例(49.4%),B组32例(37.6%),C组11例(12.9%),8岁以下儿童74例,占87.0%。男39例,女46例,男女之比为0.85∶1.00。发病年龄2个月至14岁,平均(3.90±3.39)岁,各年龄组男女比例组间比较,差异无统计学意义(P>0.05)。见图1。
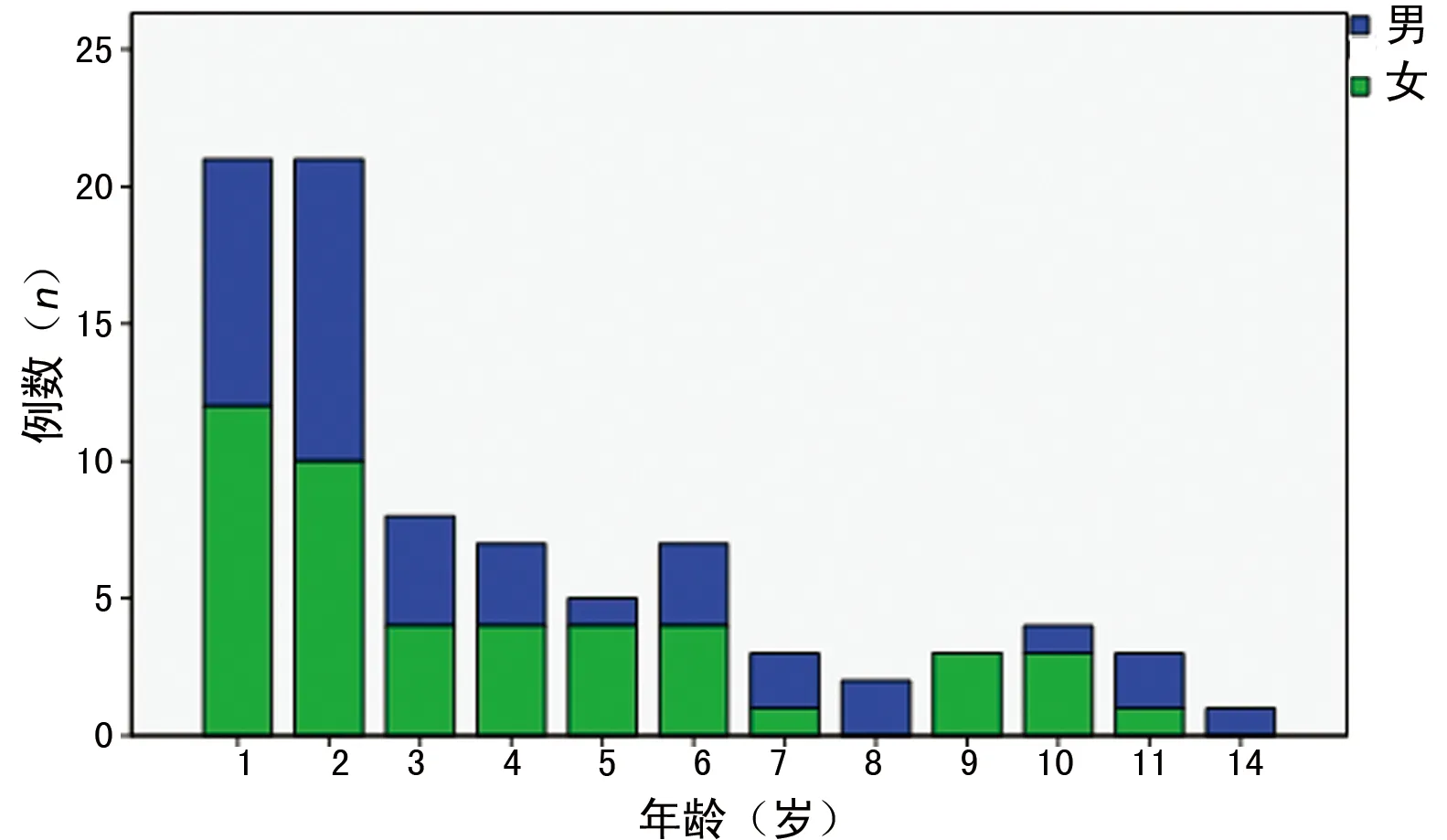
图1 85例HPS的发病年龄分布
2.2 临床表现
2.2.1 发热 85例患儿均有发热(100.0%),温度在38.5~40.5 ℃,发热时间大于或等于10 d者,在A组中有36例(85.7%),B组中有26例(81.3%),C组为11例(100.0%),组间比较,差异无统计学意义(P>0.05)。见表1。
2.2.2 肝脏肿大 为肋下2~10 cm,A、B、C组肝脏肿大患儿分别是38例(90.5%),32例(100.0%),6例(54.6%),A和C组差异有统计学意义(χ2=7.983,P<0.05),B和C组差异有统计学意义(χ2=16.459,P<0.05)。见表1。
2.2.3 脾脏肿大 表现为肋下1~10 cm,A组35例(83.3%),B组28例(87.5%),C组4例(36.4%),3组比较,A和C组差异有统计学意义(χ2=9.894,P<0.05),B和C组差异有统计学意义(χ2=11.244,P<0.05)。见表1。
2.2.4 颈部淋巴结肿大 A组20例(47.6%),B组25例(78.1%),C组5例(45.5%),C组比较,A和B组差异有统计学意义(χ2=7.092,P<0.05),B和C组差异有统计学意义(χ2=4.142,P<0.05)。见表1。
2.2.5 咽峡炎 A组发生有20例(47.6%),B组28例(87.5%),C组7例(63.6%),A和B组差异有统计学意义(χ2=12.675,P<0.05)。见表1。
2.2.6 并发肺炎 A组有23例(54.8%),B组17例(53.1%),C组3例(27.3%),3组相互比较,差异无统计学意义(P>0.05)。见表1。
2.2.7 浆膜腔积液(胸腹腔) A组有15例(35.7%),B组21例(65.6%),C组6例(54.6%),A和B组差异有统计学意义(χ2=6.504,P<0.05)。见表1。
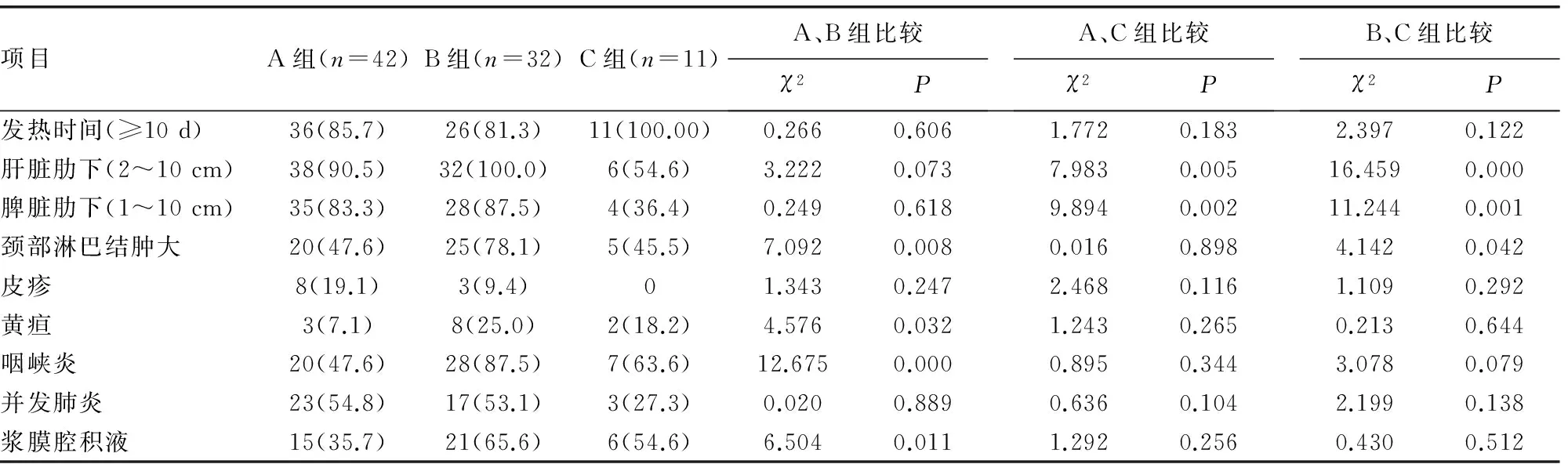
表1 儿童各年龄组HPS 临床表现[n(%)]

表2 儿童各年龄组HPS实验室检查结果及比较±s)
2.2.8 其他症状 如皮疹、黄疸的发生率均较低,皮疹A组有8例(19.1%),B组3例(9.4%),C组0例,组间差异无统计学意义(P>0.05)。黄疸A组有3例(7.1%),B组8例(25.0%),C组2例(18.2%),仅A和B组差异有统计学意义(χ2=4.576,P<0.05)。C组有1例发生中枢神经系统改变(表现为症状性癫痫,脑萎缩)。见表1。
2.2.9 病因 A组EB病毒感染15例(占35.7%)、1例家族噬血、1例继发于化脓性脑膜炎、1例继发于淋巴瘤、2例巨细胞病毒感染,病因未明22例。B组中EB病毒感染15例(占46.8%)、1例继发于川崎病,1例继发于类风湿性关节炎、1例继发于黑热病,病因未明14例。C组中有EB病毒感染8例(占72.7%)、1例继发于白血病,1例继发于川崎病,1例继发于EBV阳性的T淋巴瘤(EBV检测IgM抗体,2012年以后增加EBV-DNA检测)。其中18例患儿EBV-DNA阳性,但EBV-IgM抗体阴性。A、B、C组EBV总的感染率为44.7%。A、B组EBV感染率比较,差异无统计学意义(P>0.05);A、C组间比较差异有统计学意义(P<0.05);B、C组间比较,差异有统计学意义(P<0.05)。
2.3 实验室检查
2.3.1 血常规、生化及免疫等检查 3个年龄组的实验室检查结果分析见表2。血常规、生化及免疫等检查结果经过t检验,发现白细胞、中性粒细胞绝对数、血红蛋白、血小板、纤维蛋白原、三酰甘油(TG)、丙氨酸氨基转移酶(ALT)和天门冬氨酸氨基转移酶(AST)、细胞免疫CD4/CD8的比值在3个不同年龄组间比较,差异无统计学意义(P>0.05),仅乳酸脱氢酶(LDH)测定值在A和B组间比较,差异有统计学意义(t=-2.042,P<0.05),血清铁蛋白检查,以>500 μg/L进行统计,0~2岁组阳性率为90.0%,>2~8岁组和>8~14岁组阳性率均为100.0%,组间比较差异无统计学意义(P>0.05)。
2.3.2 骨髓检查 有核细胞表现为增生活跃者A组29例(69.1%),B组24例(75.0%),C组11例(100.0%),组间比较差异无统计学意义(P>0.05)。粒细胞红细胞比例(粒红比)小于1.0者A组13例(31.0%),B组8例(25.0%),C组0例,组间比较,A和C组差异有统计学意义(P<0.05)。噬血细胞检出率高,A组40例(95.2%),B组和C组都全部检测出(100.0%),组间比较差异无统计学意义(P>0.05)。见表3。
2.3.3 基因检查 2012年后开始对临床确诊为HPS的32例患儿进行基因检查。发现阳性病例1例,为UNC13D基因Exon27上存在错义突变,C.2588G>A;P.Gly863Asp杂合,该病例为女,1.5岁,按2004年方案正规化疗到33周时疾病再活动,后经治疗无效死亡(未行骨髓移植),其父母也发现携带相同突变基因(其父母均未发病)。
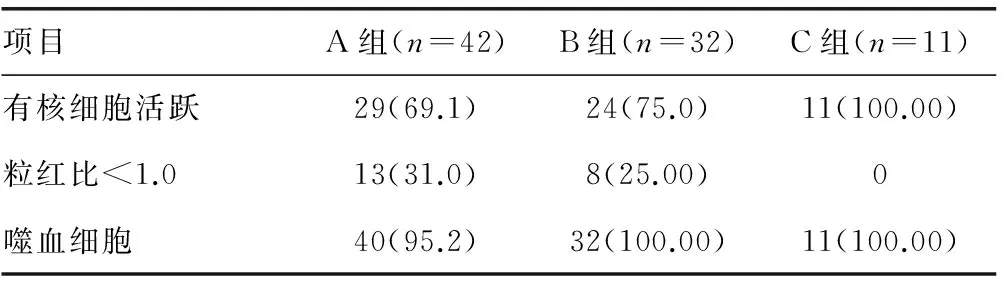
表3 HPS各年龄组骨髓检查统计结果[n(%)]
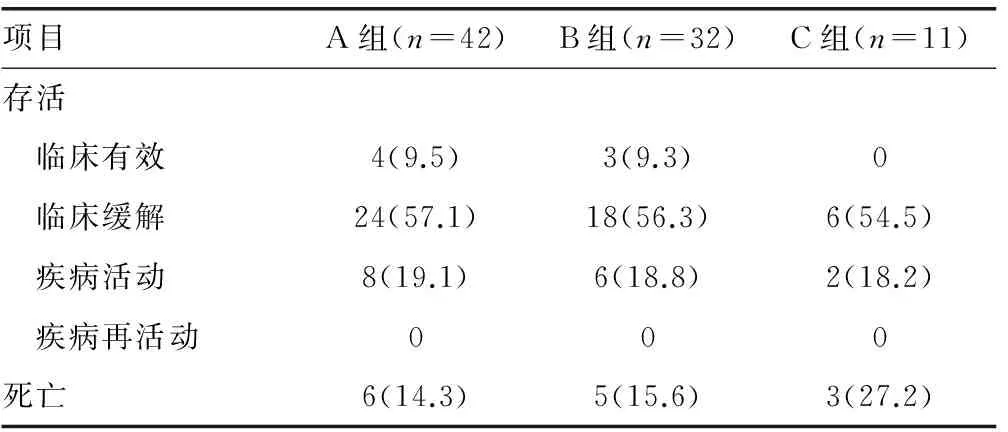
表4 85例HPS治疗转归结果分析[n(%)]

表5 HPS存活和死亡患儿临床特点比较结果
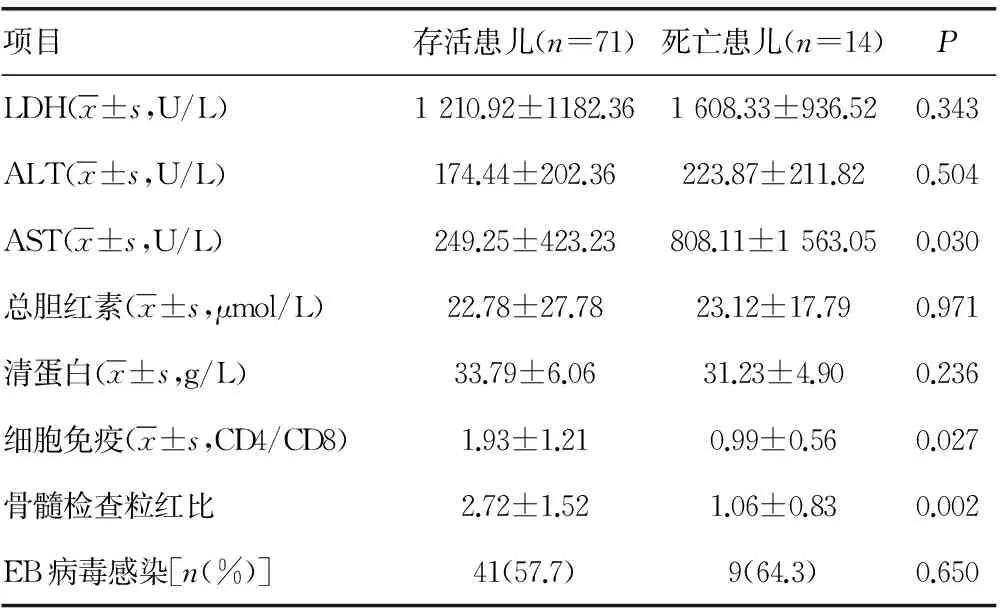
续表5 HPS存活和死亡患儿临床特点比较结果
2.4 治疗转归结果 12例患儿治疗早期进行了血浆置换,其中11例临床缓解,1例死亡。所有HPS患儿经上述治疗方案治疗后其治疗转归见表4,临床有效、临床缓解、疾病活动和疾病再活动4种转归在3个不同年龄组间差异无统计学意义(P>0.05)。存活71例(83.5%)和死亡14例(16.5%),两种转归在3个不同年龄组间比较,差异无统计学意义(P>0.05)。治疗效果佳(包括临床有效、临床缓解)的患儿A组有28例(66.7%),B组有21例(65.6%),C组有6例(54.6%),组间比较差异无统计学意义(P>0.05)。
2.5 预后因素分析
2.5.1 单因素分析 将年龄、临床表现、实验室检查各项指标在存活患儿和死亡患儿间进行比较分析,结果浆膜腔积液(χ2=7.213,P<0.05),血小板(t=2.061,P<0.05),TG(t=-3.602,P<0.05),AST(t=-2.220,P<0.05),细胞免疫(CD4/CD8)(t=2.266,P<0.05)及骨髓检查粒红比(t=3.179,P<0.05),差异有统计学意义。而存活与死亡患儿在发病年龄、肝脾肿大,白细胞计数、血红蛋白、纤维蛋白原、LDH及EB病毒感染等指标比较,差异无统计学意义(P>0.05)。见表5。
2.5.2 多因素分析 将从存活和死亡患儿对比分析差异有统计学意义的因素与HPS病死率相关的危险因素进行Logistic多因素回归分析,结果显示TG>3.5 mmol/L、CD4/CD8<1.24、骨髓粒红比小于1.02是预后不良的危险因素(P<0.05)。见表6。
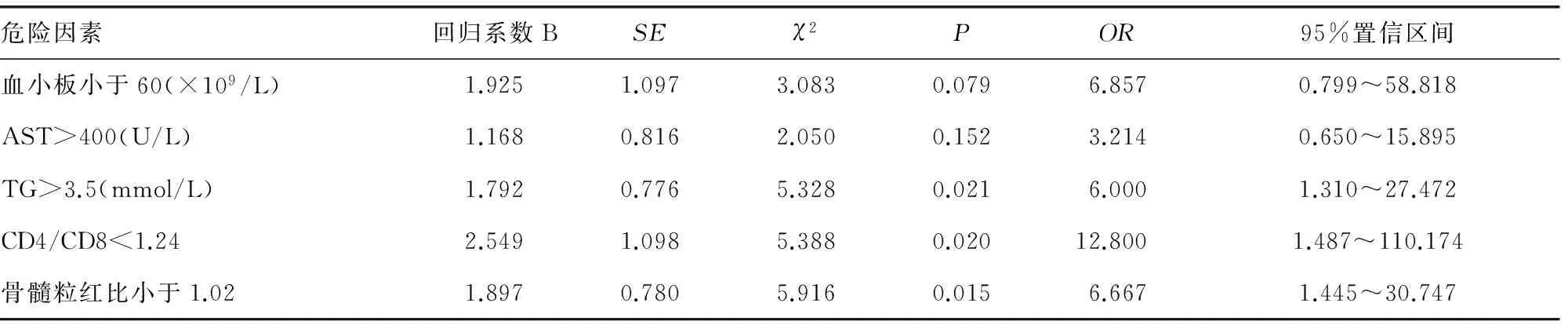
表6 HPS病死率多因素相关性分析
3 讨 论
HPS是一种多器官、多系统受累,伴免疫功能紊乱的巨噬细胞增生性疾病,其特征是持续高热,肝、脾、淋巴结肿大,凝血障碍、外周全血细胞减少,肝功能急剧恶化和中枢神经系统功能障碍等,病程凶险,病死率高。HPS不是一种独立的疾病,而是一组有共同临床表现的综合征[8]。
本组资料收集病例的时间跨度大,随访时间长,并将85例HPS患儿分为0~2岁、>2~8岁和>8~14岁3个不同年龄组,通过分析各组的临床特点,探究儿童HPS的病因、病理、临床症状、实验室检查及预后等方面在各个年龄段有无明显差异,儿童HPS不同年龄组各有特点,有部分临床特点与发病年龄有关。
HPS根据病因不同分为原发性和继发性两种[9]。原发性或称家族性噬血细胞综合征(familial hemophago-cytic lymphohistioclytosisi,FHL)为常染色体隐性遗传病(如穿孔素基因和Uncl3D基因等[10]),其发病和病情加剧常与感染有关,90%的发病儿童年龄小于2岁,病死率非常高[9];而FHL的诊断需要有阳性家族史或基因检查等确诊,故诊断困难。2012年以后临床确诊为HPS的32例患儿均进行了噬血基因检测,基因检测阳性仅有1例,女,1.5岁。该患儿化疗后期复发死亡(因经济原因未做移植)。故提醒在临床中发现基因异常的宜早做造血干细胞移植。继发性噬血细胞综合征(secondary hemophagocytic lymphohistiocytosisi,sHLH)由病毒感染、免疫介导、肿瘤等引起,其中以EB病毒感染最为多见[11]。本组病例中仍以EB病毒感染最多见,占44.7%,比文献报道56.3%~63.4%稍低[12-13]。EB病毒的阳性检出率(IgM阳性或DNA阳性)分别为A组35.7%,B组46.8%,C组72.7%,A、C组,B、C组组间比较,差异有统计学意义。考虑大于8岁儿童EB病毒感染更多见,<2岁儿童家族噬血多见。同时发现,有18例患儿EB病毒DNA为阳性,其EB-IgM为阴性,推测IgM阳性率低是否因为患儿免疫功能紊乱,产生抗体滴度较低所导致。故在临床中,同时送检EBV-IgM和EB病毒DNA,能明显提高EB病毒感染的阳性检出率。EB病毒诱发HPS的机制尚不清楚,目前认为主要是T淋巴细胞和被活化的巨噬细胞分泌大量的细胞因子引起的细胞因子“风暴”所致[14-15]。
儿童HPS的病理生理特点主要是单核巨噬细胞系统中发现良性的淋巴组织细胞浸润,组织细胞呈吞噬现象(噬血细胞),骨髓多增生活跃,粒系统所占比例降低[16]。本研究85例HPS患儿全部做了骨髓检查,在0~2岁组别中查见噬血细胞的阳性率为95.2%,其余2个组别阳性率均为100.0%,粒红比小于1.0患儿比例为24.7%(21/85),均发生在小于或等于8岁患儿。
通过对3组患儿的比较,临床表现各有特点。85例患儿年龄分布情况为0~2岁组别最多,占49.4%,8岁及以下占87.1%,分析原因与年龄小,其免疫功能尚未健全,受感染后细胞免疫易出现调节失衡有关,而且家族噬血多发生于2岁以下。发热时间差异无统计学意义,肝脾肿大、淋巴结肿大、黄疸、咽峡炎均在2~8岁患儿最多见,故在临床工作中遇到年龄较小的儿童时,病程早期EB病毒感染症状可能不突出,若持续高热合并血象两系低下,需积极进行噬血相关的实验室检查并随访症状、体征的变化,警惕HPS的发生,及时明确诊断。本组资料发现,噬血合并浆膜腔积液较多见,尤其是B、C组患儿。实验室检查观察到血常规中白细胞减少不明显,血红蛋白有降低,而血小板减少最为明显,这与文献报道一致[16]。在3个不同年龄组别中比较,血象改变差异均无统计学意义。生化指标对比观察,在3个不同年龄组中比较仅LDH在0~2岁和>2~8岁患儿间比较,差异有统计学意义(P<0.05)。3组患儿铁蛋白都出现明显升高,考虑铁蛋白是一个较为敏感的实验室指标,可作为观察临床疗效和疾病是否活动的指标。HPS临床进展快,病程凶险,对疑诊患儿须高度警惕,诊断依据尚不足时,须及时随访实验室指标,如铁蛋白、纤维蛋白原等,并积极送检NK细胞活性、可溶性CD25和噬血基因,一旦诊断明确,须按HLH-2004方案正规化疗。
儿童HPS的进展迅速、病死率高,确诊后应立即开始治疗,国际组织细胞协会先后公布有HLH-94方案、HLH-04方案,两方案同样适用于家族性和继发性 HPS中严重病例及病情持续存在的病例[16]。具体主要包括解除诱发因素如病毒和细菌的感染(主要针对继发性HPS)、支持治疗、大剂量静脉滴注丙种球蛋白及糖皮质激素免疫调节及遏制细胞因子风暴,化学治疗[依托泊苷(VP16)和环孢素A]及造血干细胞移植等。对于家族性HPS,惟一根治方法是化疗缓解后予以造血干细胞移植。但是由于家族性和继发性 HPS之间鉴别困难,化疗仍然是目前最主要的治疗方法。本组病例在2004年前在采用抗病毒、抗细菌、对症支持治疗的同时多采用激素和大剂量丙种球蛋白治疗。2004年以后根据病情加用VP16、环孢素A等化疗药物。另外还发现本组资料中有12例危重患儿在治疗早期同时实施了血浆置换,次数为3~4次,而这12例患儿的预后为11例临床缓解、1例死亡。国内外研究证实血浆置换能够降低病死率[17],改善难治性HPS的病情,提高其生存率。血浆置换可能机制为:(1)清除致病因子;(2)排除异常血浆成分,如炎性细胞因子、炎性介质等;(3)补充血浆因子,如清蛋白、凝血因子及其他生物活性因子等;(4)免疫调节作用,去除细胞及体液免疫抑制因子,恢复免疫功能,促进T细胞亚群恢复正常比例[18]。本组病例临床有效和缓解的3组患儿分别为66.7%、65.6%、54.6%,差异无统计学意义(P>0.05)。3组病例存活和死亡两种转归差异无统计学意义(P>0.05)。
由于儿童HPS临床过程进展迅速,预后不良,国内众多学者试图从儿童HPS的临床特点和预后结果中寻求关联因素,找出预后不好的危险因素。刘瑞海等[19]在2004年通过对32例小儿HPS的临床特点及死亡因素分析发现血小板小于20×109/L、CD4/CD8<1.160、年龄小于2岁是与死亡有关的危险因素。叶璟[20]在2005年通过对25例小儿HPS预后相关因素分析指出EB病毒感染、CD4/CD8比例倒置、血小板降低为死亡危险因素。2009年刘苗等[21]在对儿童HPS 28例临床特点和预后分析中发现年龄小于2岁、LDH>2 000 U/L是预后危险因素。2013年王华等[22]在54例儿童HPS临床及预后因素分析中提出EB病毒感染、血小板降低、ALT水平升高是死亡的独立危险因素。张雯雯等[23]在2015年通过回顾分析72例小儿HPS的发病机制及预后危险因素发现年龄小于3岁、LDH>2 500 U/L是预后不良的危险因素。而本研究通过对85例HPS患儿进行不同年龄组别的临床特点和预后因素分析,发现TG>3.5 mmol/L、CD4/CD8<1.24、骨髓粒红比小于1.02是预后不良的危险因素。结果与其他学者比较,仅CD4/CD8比例倒置相同,而其他危险因素结果却不尽相同。分析原因可能是因为儿童HPS为少发病,各组样本量较小。建议国内通过建立儿童HPS的联合研究中心,收集大宗的病例,分析大数据以得出比较准确的预后分析,有利于尽早的有针对性的治疗干预,从而降低儿童HPS的病死率。
[1]张之南,沈悌.血液病诊断及疗效标准[M].3版.北京:科学出版社,2007:254-256.
[2]Janka GE.Familial and acquired hemophagocytic lymphohistiocytosis[J].Eur J Pediatr,2007,166(2):95-109.
[3]Henter JI,Elinder G,Ost A.Diagnostic guidelines for hemophagocytic lymphohistiocytosis.The FHL Study Group of the Histiocyte Society[J].Semin Oncol,1991,18(1):29-33.
[4]Henter JI,Samuelsson-Horne A,Arico M,et al.Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation[J].Blood,2002,100(7):2367-2373.
[5]Henter JI,Horne A,Arico M,et al.HLH-2004:Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis[J].Pediatr Blood Cancer,2007,48(2):124-131.
[6]Janka GE,Schneider EM.Modern management of children with hemophagocytic lymphohistiocytosis[J].Br J Haematol,2004,124(1):4-14.
[7]Verbsky JW,Grossman WJ.Hemophagocytic lymphohistiocytosis:diagnosis,pathophysiology,treatment,and future perspectives[J].Ann Med,2006,38(1):20-31.
[8]Rouphael NG,Talati NJ,Vaughan C,et al.Infections associated with hemophagocytic syndrome[J].Lancet Infec DIS,2007,7(8):814-822.
[9]Janka GE.Familial and acquired hemophagocytic lymphohisiocytosis[J].Eur J Pediatr,2007,166(1):95-109.
[10]Horne A,Ramme KG,Rudd E,et al.Characterization of PRF1,STX11 and UNC13D genotype-phenotype correlations in familial hemophagocytic lymphohistiocytosis[J].Br J Haematol,2008,143(1):75-83.
[11]Lee WI,Chen SH,Hung IJ,et al.Clinical aspects,immunologic assessment,and genetic analysis in Taiwanese children with hemophagocytic lymphohistiocytosis[J].Pediatr infect Dis J,2009,28(1):30-34.
[12]于润红,曾利,邹旭凤,等.儿童病毒相关性噬血细胞综合征32例临床研究[J].中国小儿血液与肿瘤杂志,2010,15(1):11-17.
[13]卢愿,金润铭,吕淑华,等.噬血细胞综合征22例[J].实用儿科临床杂志,2008,23(15):1183-1186.
[14]ShimadaA,KatoM,TamuraK,etal.
Hemophagocytic lymphohistiocytosis associated with uncontrolled inflammatory cytokinemia and chemokinema was caused by systemic anaplastic large cell lymphoma:a case report and review of the literature[J].J Pediatr Hematol Oncol,2008,30(10):785-787.
[15]Areei RJ.When T cells and macrophages do not talk:the hemophagocyticsyndromes[J].Curr Opin Hematol,2008,15(4):359-367.
[16]盛光耀.儿童噬血细胞综合征的诊断与治疗[J].实用儿科临床杂志,2008,23(3):161-163.
[17]Busund R,Koukline V,Utrobin U,et al.Plasmapheresis in severe sepsis and septic shock:a prospective,randomised,controlled trial[J].Int Care Med,2002,28(10):1434-1439.
[18]陈涛,姚亚洲,郑引索.血浆置换联合化疗对噬血细胞综合征疗效观察[J].西北国防医学杂志,2012,33(3):239-241.
[19]刘瑞海,刘华林,梁卉,等.32例小儿噬血细胞综合征临床特点及死亡危险因素分析[J].中国小儿血液与肿瘤杂志,2004,9(4):163-166.
[20]叶璟.小儿噬血细胞综合征预后相关因素分析[J].中国小儿血液与肿瘤杂志,2005,10(2):54-56.
[21]刘苗,徐佳伟,王艳荣,等.儿童噬血细胞综合征28例临床特点和预后分析[J].临床儿科杂志,2009,27(8):727-730.
[22]王华,高文瑾,刘安生,等.儿童噬血细胞综合征54例临床及预后因素分析[J].陕西医学杂志,2013,18(9):31-34.
[23]张雯雯,陈红兵,朱君,等.小儿噬血细胞综合征的发病机制及预后危险因素的分析[J].南京医科大学学报,2015,35(8):1118-1122.
Characteristics and prognosis of child hemophagocytic syndrome in different age groups
LiHui,LiDan,HeYulei,ShiHong,ZhouMin△
(DepartmentofHematologyOncology,ChengduMunicipalWomenandChildrenCentralHospital,Chengdu,Sichuan610091,China)
Objective To explore the clinical characteristics and prognosis factors of child hemophagocytic syndrome(HPS) in different age groups.Methods Eighty-five children cases of HPS were divided into 3 age groups,0-2 years old (group A),>2-8 years old (group B) and >8-14 years old(group C).The etiology,pathology,clinical symptoms,laboratory detection and prognosis were retrospectively analyzed.Results In the etiology,the etiology in the group A,B and C was dominated by EB virus infection,the infection positive rate in the group C was significantly higher than that in the group B and C;in the bone marrow examination:the occurrence rate of granulocytes to erythrocytes ratio <1.0 in the group A was higher than that in the group C;in the clinical symptoms,cervical lymph node enlargement mostly occurred in the group B,hepatosplenomegaly was most rare in the group C,and the serous cavity effusion commonly occurred in the group B;in the laboratory detection,the LDH level in the group B was higher than that in the group A,the above differences were statistically significant(P<0.05).There were no statistically significant differences in other clinical symptoms and laboratory detection results among the three groups (P>0.05);In the comparison between the survival group and death group,the serous cavity effusion,triglyceride(TG),CD4/CD8 ratio and granulocytes to erythrocytes ratio in bone marrow had statistical difference(P<0.05).The above factors were performed the Logistic analysis,the results showed that TG>3.5 mmol/L,CD4/CD8 ratio <1.24 and bone marrow granulocytes to erythrocytes ratio <1.02 were the unfavorable risk factors affecting the prognosis of child HPS (P<0.05).Conclusion Partial clinical characteristics of child HPS are related with the onset age,TG level,CD4/CD8 ratio and bone marrow granulocytes to erythrocytes ratio are the risk factors affecting prognosis.
lymphohistiocytosis;hemophagocytic;age factors;clinical characteristics;prognosis;risk factor
李蕙(1974-),副主任医师,本科,主要从事血液肿瘤的研究。△
,E-mail:zhoumin7266@126.com。
10.3969/j.issn.1671-8348.2017.22.022
R725.5
A
1671-8348(2017)22-3092-05
2017-01-18
2017-03-28)
