分子动力学模拟研究矿物基础油润滑性能
2017-08-31李义雅段庆华代振宇
李义雅, 龙 军, 段庆华, 代振宇, 赵 毅, 苏 朔
(中国石化 石油化工科学研究院, 北京 100083)
分子动力学模拟研究矿物基础油润滑性能
李义雅, 龙 军, 段庆华, 代振宇, 赵 毅, 苏 朔
(中国石化 石油化工科学研究院, 北京 100083)
采用分子动力学(MD)模拟方法,以十四烷基环己烷为模型化合物,主要从烃分子与Fe(110)表面的吸附能以及烃分子之间的作用能2个方面,研究了矿物基础油分子在Fe(110)表面的润滑作用,并比较了烃分子结构及温度对润滑作用的影响。结果表明,烃分子与Fe(110)表面的吸附能远大于烃分子之间的作用能,因而能形成稳定的润滑油膜,这2种作用能主要来源于范德华作用。稳定润滑状态下,烃分子水平分布的吸附能比竖直分布的大,更容易水平分布在Fe(110)表面。不同结构烃分子之间的作用能不同,其由大到小顺序为:芳香烃,环烷烃,直链烷烃,异构烷烃;不同结构烃分子与Fe(110)表面的吸附能也不相同,其由大到小顺序为:芳香烃,直链烷烃,环烷烃,异构烷烃;这2种作用能均随温度的升高而降低,使润滑油膜的稳定性下降。
基础油;润滑性能;分子动力学模拟;吸附能;分子间作用能
固体表面不是绝对平整光滑的,即使经过精密加工的机械零件表面也存在许多微小的凸起和凹谷。当表面作相对运动时,固体表面要克服金属表面间的黏附作用,又要克服粗糙微凸体引起的机械变形阻力,从而产生摩擦[1]。伴随着摩擦还会产生多种形式的磨损,引起机械部件的损坏。据估计,世界上一次能源大约1/3消耗于摩擦,约有80%的设备损坏是由于各种形式的磨损引起的[2]。为减少摩擦磨损带来的经济损失,最大限度地减少摩擦阻力、降低机械磨损是一项社会经济效益课题。目前,改善摩擦磨损的主要形式有材料加工、表面改性和薄膜技术[3],薄膜技术包含固体膜、半固体膜、液体膜和气体膜,液体膜中最重要也最常用的是润滑油,润滑油大约占总润滑材料的85%。
润滑油可以在金属表面间形成一层稳定的保护膜,避免2个金属表面的直接接触,从而发挥减摩抗磨作用。用润滑油层较小的内摩擦代替摩擦系数较大的金属表面摩擦,发挥减摩作用。润滑油由基础油和多种功能添加剂组成,基础油是其主要组分,占润滑油成分的90%以上,决定润滑油的基本性质。基础油主要分为矿物基础油、合成基础油和生物基础油[4],其中矿物基础油的应用最为广泛,大约占95%以上。
矿物基础油的主要组成为C20~C40的直链烷烃、异构烷烃、环烷烃和芳香烃。润滑油发挥润滑作用的关键是形成一层稳定的保护膜,保护膜的稳定性与2个因素有关,一是保护膜分子与金属表面间的吸附作用,另一个是保护膜体相分子之间的作用,这2个作用越强,保护膜越稳定。同时,若要发挥减摩作用,保护膜的内摩擦力也应较低,内摩擦力的大小也与分子间作用能相关。目前,对润滑性能的研究,主要是通过摩擦试验机和四球机等模拟试验,得到摩擦系数、磨斑直径和最大无卡咬负荷等性能参数,以此来说明润滑油的减摩抗磨效果[5-9]。而这些方法难以研究微观的吸附作用和分子间作用,因此需要用新的方法来研究分子水平的本质作用。
分子模拟是随着量子化学理论和计算机技术进步而发展的一种计算化学方法,可以研究物质在原子分子水平的物理和化学性质。该方法作为一种重要的研究手段,在矿物基础油研究领域已经有了一定应用。Jabbarzadeh等[10]用分子动力学研究C30烷烃支化度对润滑膜的影响,结果表明,支化度越高,分子越不容易成膜,黏度也越高,并且表面剪切速率增高时,直链分子的黏度降低较多,而高支化度分子的黏度没有明显变化。Liu等[11]用非平衡分子动力学方法计算了润滑油的剪切黏度与温度和压力的关系,压力/黏度系数符合实验测得的数据,可以用该系数预测基础油黏度与压力的关系。但是,目前对矿物基础油润滑作用的机理还缺乏深入细致的研究。笔者采用分子动力学方法,以含有20个碳原子的十四烷基环己烷以及直链烷烃、异构烷烃、环烷烃和芳香烃为模型化合物,研究矿物基础油在Fe(110) 表面间的润滑作用机理,分析烃分子在润滑平衡态下的构型,考察温度和分子结构对矿物基础油润滑作用的影响。
1 分子动力学计算方法
1.1 计算模型和参数
矿物基础油的理想组分是少环长侧链的烃类,其碳数分布在C20~C40。为提高计算效率,选择含有20个碳原子的十四烷基环己烷为模型化合物研究矿物基础油的润滑性能,其结构如图1(a)所示。为比较矿物基础油中不同分子结构的影响,选择C20的4种结构为模型化合物,分别为直链烷烃、异构烷烃、环烷烃和芳香烃,结构如图1(b)~1(e)所示。
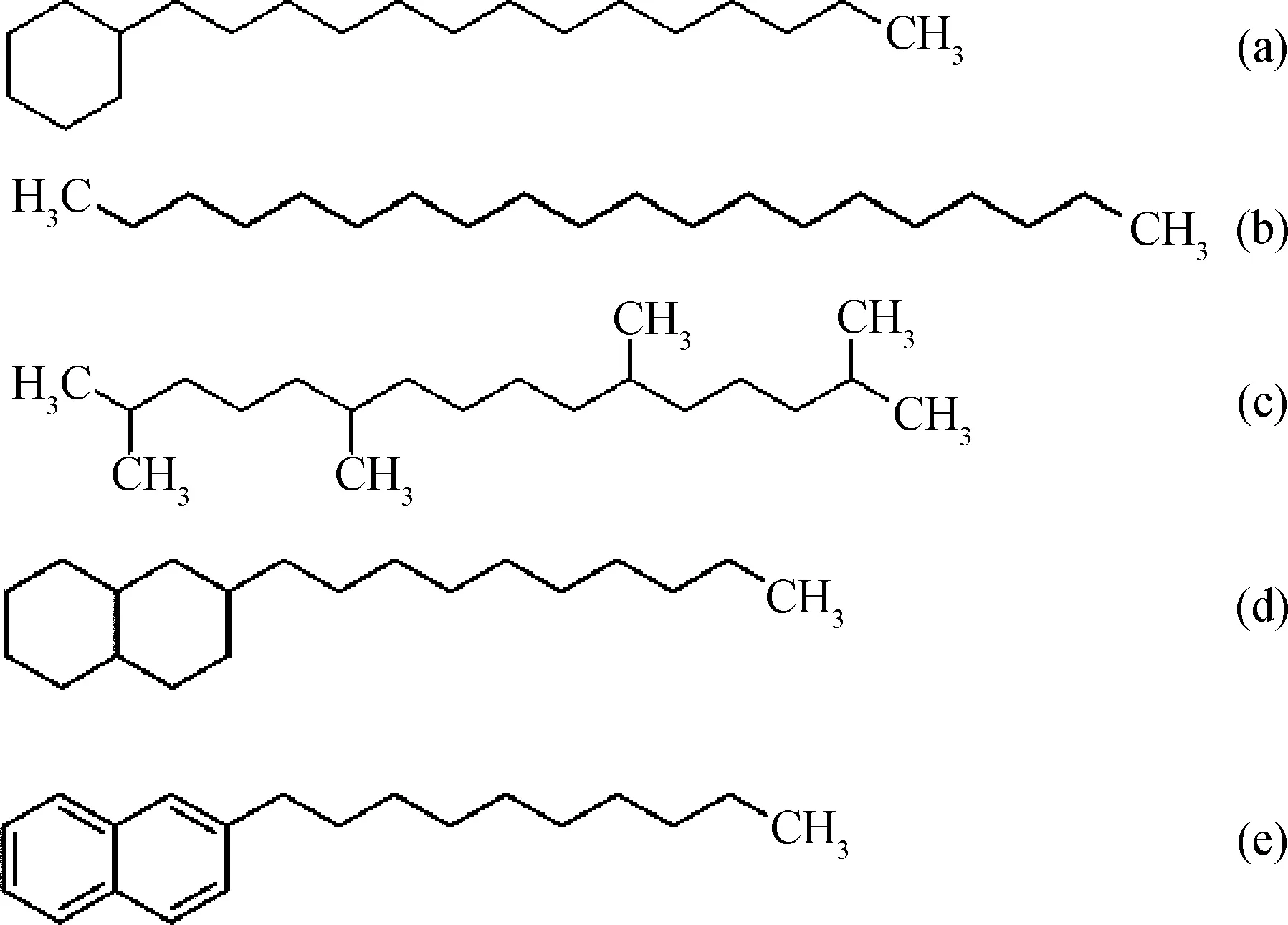
图1 矿物基础油的分子结构Fig.1 Molecular structure of mineral base oil (a) Tetradecylcyclohexane; (b) n-Eicosane;(c) 2,6,11,15-Tetramethyl hexadecane;(d) 2-Decyl decahydronaphthalene; (e) 2-Decyl naphthalene
由于Fe(110)表面是生长速率最慢的表面,在Fe晶体中所占比例最高,选择Fe(110)表面为金属表面。构建表面体系的大小为2.9789 nm×2.9789 nm×1.2245 nm,厚度为7层,表面层俯视图如图2(a)所示,侧视图如图2(b)所示。

图2 Fe(110)表面模型Fig.2 The model of Fe(110) surfaces (a) Vertical view; (b) Side view
计算过程采用Materials Studio 8.0中的Forcite模块,力场为COMPASSⅡ,静电作用和范德华作用的非键截断均采用Ewald算法。分子动力学模拟过程中,计算的时间步长均为1.0×10-15s,有关压力和温度的控制函数分别采用Berendsen和Nose方法。
1.2 模拟过程
先用几何优化和分子动力学平衡找到十四烷基环己烷分子的最低能量构象,以该构象为计算模型,采用Amorphous Cell建立含有50个分子、密度为0.4 g/mL的无定形晶胞。用等温等压系综(NPT)在20℃下,对该晶胞进行退火及动力学平衡计算,平衡后统计计算得到该温度下晶胞的密度为0.8107 g/mL,实验值为0.8258 g/mL,两者相近,说明选择的参数合理。采用同样的方法,计算全部5种结构基础油分子在25℃下的密度,分别为0.8201、0.7791、0.7820、0.8526、0.9157 g/mL。
采用Build Layers工具,以上述构建的 Fe(110)表面为层,搭建含有上下2个Fe表面、中间有2.5 nm真空层的模型。在该模型上添加Connolly表面,采用Amorphous Cell的Packing方法,按照上述计算的密度,在真空层添加5种不同结构的基础油分子。为避免2个周期性结构之间作用的影响,在Fe(110)上表面添加3 nm的真空层,最终搭建的润滑模型如图3所示。
对于十四烷基环己烷分子搭建的润滑模型,在25℃下,用微正则系综(NVT)对分子层作模拟时间为2.0×10-10s的动力学弛豫,使体系充分平衡。然后用受限剪切(Confined Shear)方法,上下2个Fe(110)表面为限定层,沿x轴方向相对运动,中间的烃分子层作剪切运动,总模拟时间为1.5×10-9s。对于其它4种基础油分子搭建的模型,为考察温度对润滑油膜稳定性的影响,分别在25、75、100、150、200、250、300、350、400℃条件下,用NVT系综对烃分子层作模拟时间为2.0×10-10s的动力学弛豫。对平衡后的体系,计算烃分子层与Fe(110)表面的吸附能,以及烃分子之间的作用能。
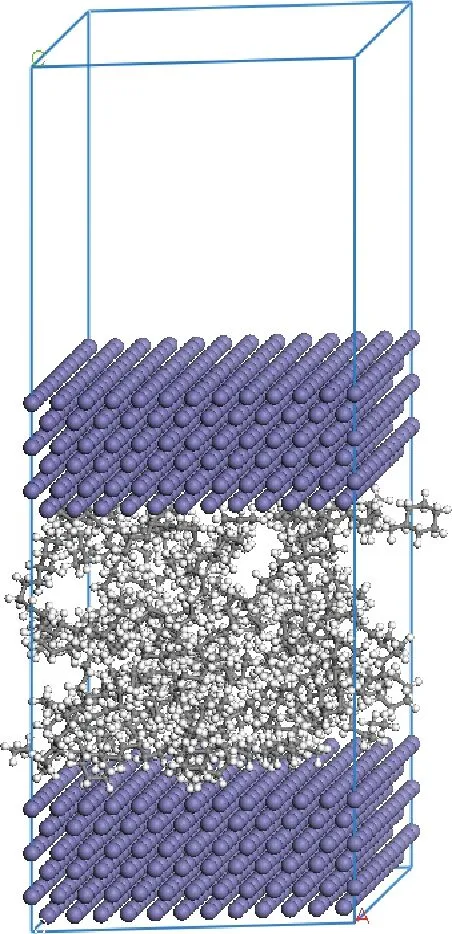
图3 润滑体系模型Fig.3 The model of the lubricant system
不同结构分子与Fe(110)表面的吸附能可通过公式(1)计算,分子间作用能用Cohesive Energy Density计算,其分子间作用能的定义为公式(2)。
Eads=Etotal,1-(Emolecule+Esurface)
(1)
Einter=Etotal,2-Eintra
(2)
式(1)、(2)中,Eads为烃分子层与Fe(110)表面的吸附能,Etotal,1为润滑模型总能量,Emolecule为烃分子层的能量,Esurface为无烃分子时Fe(110)表面的能量;Einter为烃分子层的分子间作用能,Etotal,2为烃分子层总能量,与Emolecule代表的意义相同,计算方法不同,Emolecule是用Energy计算得到的,Etotal,2是计算体系的内聚能密度(CED)时得到的,Eintra为烃分子的内能,单位均为kJ/mol。
2 结果与讨论
2.1 十四烷基环己烷的润滑作用
采用受限剪切模拟十四烷基环己烷在Fe(110)表面间的剪切运动,可观察烃分子在表面间的微观运动。分析分子在表面间的浓度分布,计算吸附能和分子间作用能的大小和具体组成,并比较剪切运动对这些参数的影响,可以得到烃分子发挥润滑作用的本质来源,深入认识矿物基础油的润滑作用。
图4为受限剪切过程中不同时间下润滑体系的构型。由图4可以观察到,当上下Fe(110)表面沿水平方向相对运动时,烃分子在水平方向作剪切运动,体系的摩擦为烃分子之间的摩擦。计算剪切前后基础油分子沿竖直方向在Fe(110)表面间的相对浓度分布,结果如图5所示。由图5可知,分子的浓度在润滑模型竖直方向的1.2 nm和3.7 nm附近最高,两者之间位置的浓度相对较低,并且剪切前后的浓度变化不大。根据搭建模型的大小,1.2 nm和3.7 nm附近是模型中Fe(110)表面的下层表面和上层表面,两者之间为烃分子。由此可以得到,在剪切运动前后,烃分子在上下Fe(110)表面的浓度均远大于体相中的浓度。说明烃分子可以稳定吸附在Fe(110)表面,并且剪切运动不会破坏这种吸附作用,运动过程中能够用烃分子较小的分子内摩擦代替较大的Fe(110)表面间摩擦,发挥减摩作用。
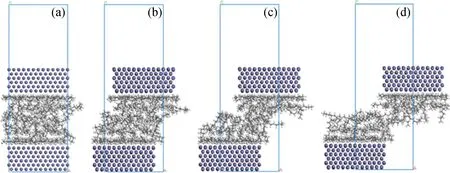
图4 不同模拟时间(tm)下润滑体系的构型Fig.4 The configuration of lubricant system at different simulation time (tm)tm/s: (a) 0; (b) 5.0×10-10; (c) 1.0×10-9; (d) 1.5×10-9

图5 剪切前后十四烷基环己烷在Fe(110)表面间的相对浓度分布Fig.5 Relative concentration distribution of Fe(110) surfaces of tetradecylcyclohexane before and after shearing
为研究十四烷基环己烷分子与Fe(110)表面的吸附能及烃分子之间的作用能本质,分析了2种作用能的大小及组成,结果如表1所示。吸附及分子间聚集过程都是放热过程,因此数值均为负值,负值越小说明吸附和分子间聚集越容易发生,即其绝对值越大吸附能和分子间作用能也越大。由此可见,吸附能均远大于分子间的作用能,说明烃分子能够克服分子间作用的阻碍,运动扩散到Fe(110)表面,稳定吸附在Fe(110)表面上,也说明分子间的作用较难使吸附在表面上的分子脱附。同时由于分子间存在一定作用能,体相基础油分子可以稳定存在于Fe(110)表面之间,使润滑油膜保持一定厚度,发挥润滑作用。由能量的具体组成可以得到,十四烷基环己烷分子与Fe(110)表面的吸附能,以及烃分子之间的作用能均主要是范德华作用,静电作用很小。这是因为选用的模型化合物为环烷烃,极性较弱,主要是范德华作用;由于矿物基础油的组成主要为各种烃类分子,极性均较弱,可以说明基础油分子形成稳定润滑油膜的作用能主要来源于范德华作用。
比较剪切对吸附能和分子间作用能的影响,剪切之后分子间作用能稍有增加,吸附能几乎没有变化。根据上述分析,这2种作用能主要来源于范德华作用,范德华作用力主要包括取向力、诱导力和色散力,由于烃分子的极性很弱,其作用主要是色散力。对于分子间作用能,处于体相的中间层烃分子会随Fe(110)表面的运动作剪切运动,剪切力使分子的正负电荷中心不重合,分子瞬时偶极矩增加,极性增加,因此色散力增加,使其分子间作用能增加。对于烃分子与Fe(110)表面的吸附能,由于Fe(110)表面相对运动时,吸附在表面的烃分子几乎没有相对运动,极性变化不大,其色散力没有明显增加,因此吸附能几乎不变。
2.2 十四烷基环己烷的稳态构型
矿物基础油通过范德华作用在金属表面间形成稳定的润滑油膜,金属表面作相对运动时,烃分子可能水平吸附于金属表面,也可能竖直吸附于金属表面。为研究基础油分子在稳态时的吸附构型,同样以十四烷基环己烷为模型化合物,分别以水平和竖直构型放置同样数量的分子在Fe(110)表面,搭建如图6(a)和6(c)的模型,在25、100、200℃条件下作动力学平衡,让分子达到在此条件下的平衡态,并计算吸附能,比较稳定性高低。

表1 剪切前后十四烷基环己烷的吸附能及分子间作用能Table 1 The adsorption energy and intermolecular interaction energy of tetradecylcyclohexane before and after shearing
EvdW—The van der Waals interaction energy among molecules;Eelect—The electrostatic effect energy among molecules
图6(b)和6(d)分别为水平和竖直构型在25℃下平衡后的形态。可以观察到,水平构型在平衡后由于分子间作用力的影响,分子堆积得更加紧密;竖直构型在平衡后分子倾斜,倾向于向水平构型分布,可直观说明烃分子更容易水平分布在金属表面。为进一步定量说明2种构型的稳定性,比较2种构型平衡后在不同温度下的吸附能,结果如图7所示。在3种温度下,水平构型的吸附能均比竖直构型的吸附能大。因此,由稳态构型和吸附能数据可以说明,烃分子水平吸附在金属表面时更稳定,在平衡润滑状态下,基础油分子更容易水平分布在金属表面。分析其原因,根据上述计算,烃分子与金属表面的吸附作用主要是范德华作用力,范德华作用与相互作用对象之间的作用位点有关,作用位点越多,范德华作用力越大。基础油分子水平分布时,分子中的所有原子均可充分与金属表面接触,作用位点多,范德华作用力大,因此吸附能大,构型更稳定。竖直分布时,由于分子间不可避免存在空隙,作用位点较少,范德华作用力小,因此吸附能小,构型稳定性较差。
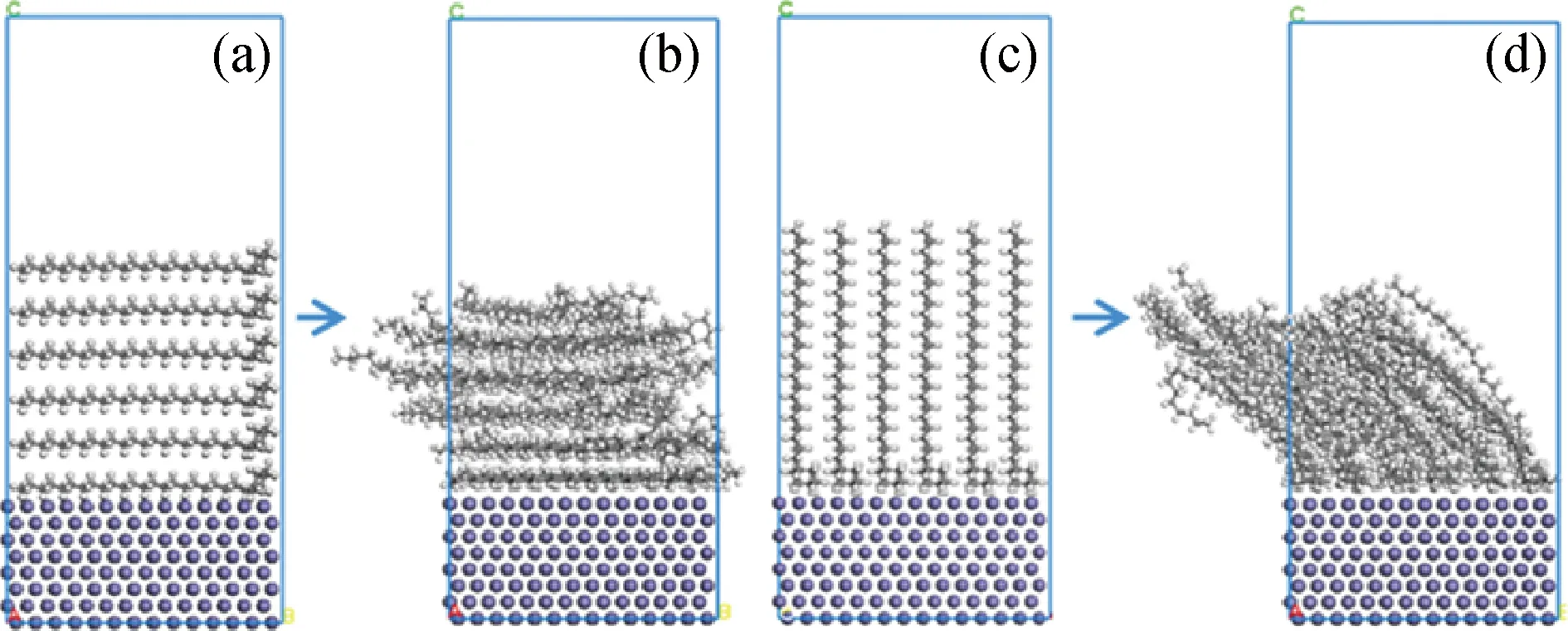
图6 平衡前后十四烷基环己烷水平和竖直分布的构型Fig.6 The horizontal and vertical configurations of tetradecylcyclohexane before and after equilibrating(a) Initial horizontal configuration;(b) Equalized horizontal configuration;(c) Initial vertical configuration; (d) Equalized vertical configuration
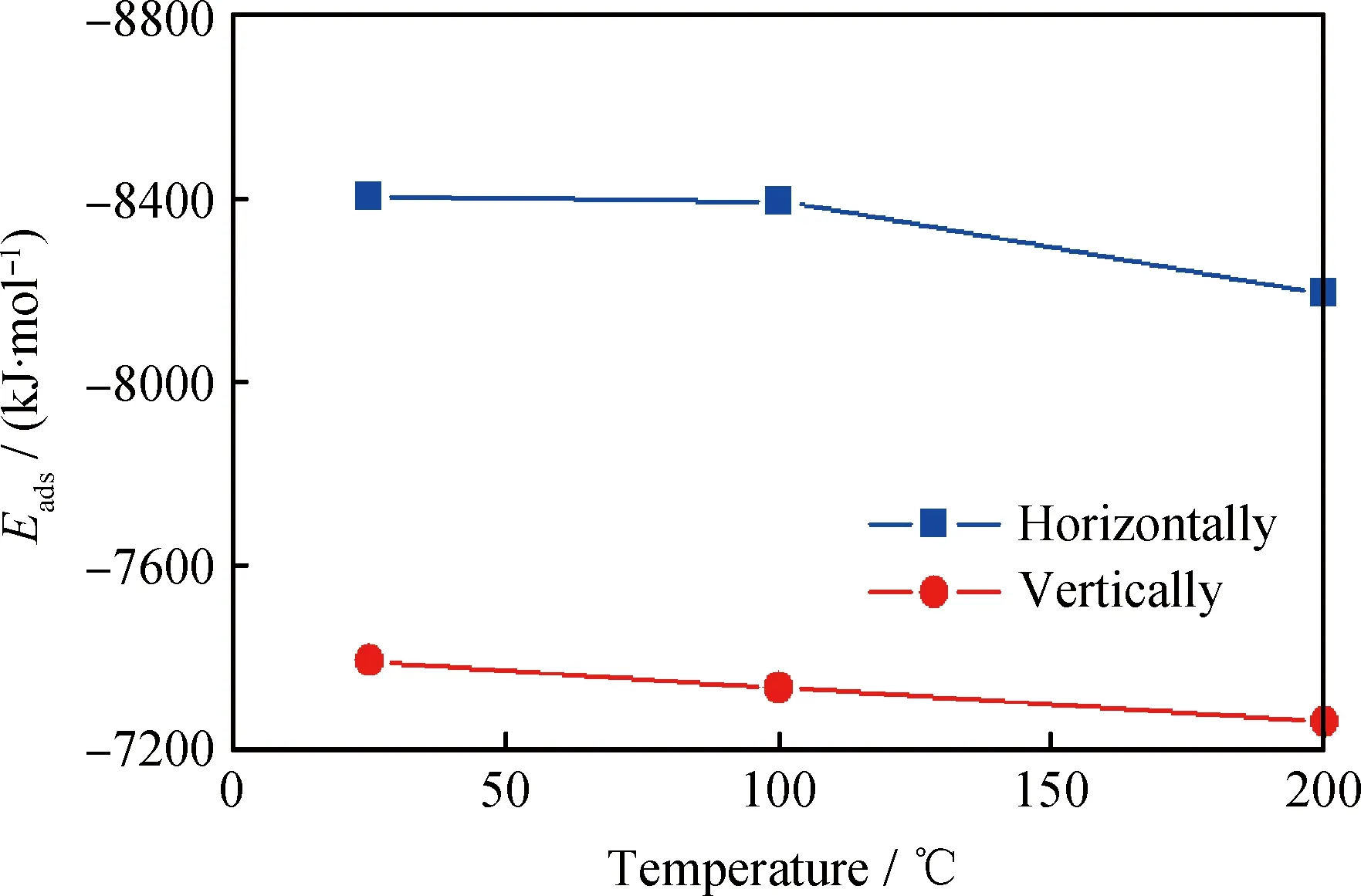
图7 水平和竖直构型在不同温度下的吸附能(Eads)Fig.7 The adsorption energy (Eads) of horizontally and vertically configurations at different temperatures
2.3 分子结构及温度对矿物基础油润滑性的影响
矿物基础油由异构烷烃、环烷烃和芳香烃组成,基础油的润滑性能也是各种烃分子润滑性能的体现。不同结构烃分子的性能不同,对基础油整体润滑性能的影响也不相同,但具体如何影响尚不清楚。研究不同分子结构的润滑性能,明确分子结构对基础油润滑性能的影响,对开发性能优良的基础油有重要的意义。另一方面,温度对基础油的润滑作用有重要影响,一般随着温度升高,基础油形成的油膜失效,润滑作用减弱。根据上述分析,基础油膜的稳定性与基础油分子与金属表面的吸附能以及基础油分子之间的作用能有关。通过研究温度对这2种作用的影响,可以从本质上认识温度对基础油润滑性能的影响。
分子结构及温度对基础油分子之间作用能的影响如图8(a)所示,不同结构烃分子之间的作用能不同,其由大到小顺序为:芳香烃,环烷烃,直链烷烃,异构烷烃。分子结构对这2种作用能的影响不同,链烷烃分子的极性小,分子间的范德华作用力弱,因此直链烷烃和异构烷烃的分子间作用力较低;同时与直链烷烃相比,异构烷烃分子链上的取代基使分子间距离增大,范德华作用降低,则异构烷烃的分子间作用力最低。环烷烃与直链烷烃相比,原子排列更紧密,分子间距离更近,分子间的范德华作用力也更强。芳香烃的极性相对最大,且原子排列紧密,因此分子间范德华作用力最强。并且随着温度升高,4种分子的分子间作用力均下降。这是因为温度升高时,分子的动能增大,分子间距离增大,范德华作用力降低。因此,温度升高会通过降低分子间作用力,降低基础油形成油膜的稳定性,从而使润滑作用降低。
分子结构及温度对基础油分子与Fe(110)表面间吸附能的影响如图8(b)所示。可以看到,不同结构烃分子与Fe(110)表面的吸附能不同,其由大到小顺序为:芳香烃,直链烷烃,环烷烃,异构烷烃。与分子间作用力不同,烃分子与Fe(110)表面的吸附能是分子与刚性平面的作用,与分子的极性和分子与Fe(110)表面的接触位点多少有关。直链烷烃的极性较小,但是其为“线型”结构,能够充分与Fe(110)表面接触,吸附能较大。而异构烷烃由于存在取代基,对分子与Fe(110)表面的接触有阻碍作用,并且其极性较低,吸附能最低。环烷烃分子为椅式或船式等“折叠”型结构,也会阻碍原子与Fe(110)表面的接触,吸附能较低。芳香烃的极性最大,并且芳香烃的苯环为“平面”结构,可使分子与Fe(110)表面充分接触,因此其与Fe(110)表面的吸附能最大。同样,随着温度升高,由于分子的动能增加,分子与Fe(110)表面间的距离减小,两者间的吸附能降低。所以,温度也可以通过降低基础油分子与Fe(110)表面的吸附能,降低基础油分子形成润滑油膜的稳定性,从而使基础油的润滑性能降低。
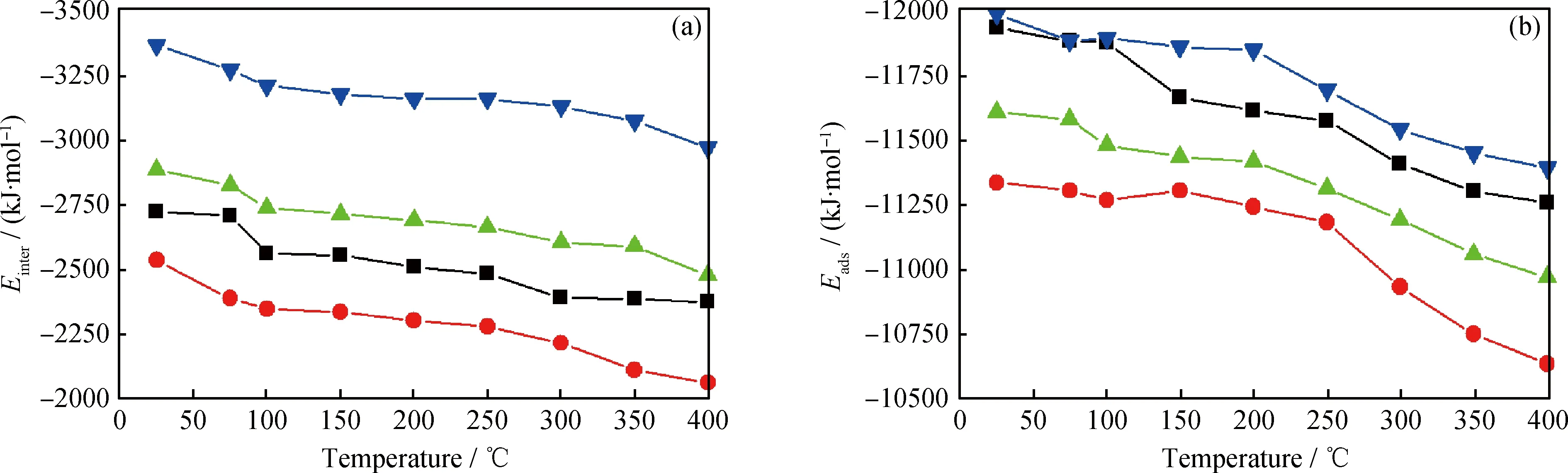
图8 分子结构和温度对分子间作用能(Einter)及吸附能(Eads)的影响Fig.8 The influence of molecular structure and temperature on intermolecular interaction energy (Einter) and adsorption energy (Eads)(a) Einter; (b) Eads Linear alkane; Isoalkane; Cylcoalkane; Aromatics
比较4种结构基础油分子的吸附能与分子间作用能可以得到,所有基础油分子的吸附能均远大于分子间作用能,说明基础油分子可以突破分子间作用的阻碍运动扩散并吸附在Fe(110)表面,分子间作用能使分子层具有一定的强度和厚度,从而形成稳定的润滑油膜,发挥减摩抗磨作用。减摩作用的效果与油膜的稳定性和内摩擦力有关。综合分析4种结构分子吸附能与分子间作用能的大小,芳香烃的吸附能和分子间作用能均最大,形成的润滑油膜最稳定。但润滑油膜的分子间作用能过大,会使分子相互运动时的阻力增大,即摩擦系数增大,从而使减摩效果降低。多分支异构烷烃的2种作用能均最低,形成的润滑油膜稳定性最差,即不能确保润滑油膜可以稳定存在于金属表面之间,较难发挥良好的润滑作用。直链烷烃和长直侧链环烷烃的吸附能和分子间作用能适中,可以保证形成稳定的润滑油膜及较低的运动阻力,但直链烷烃容易结晶会使基础油的凝点较高,不是理想的基础油组分。因此,通过比较不同结构基础油分子的吸附能和分子间作用能,可以得到长直侧链环烷烃是理想的基础油组分。在开发性能优异基础油组分时,也可以通过分析分子的吸附能及分子间作用能来预测其润滑性能。
3 结 论
(1)基础油分子形成稳定润滑油膜的作用主要来源于范德华作用,且基础油分子与Fe(110)表面的吸附能均远大于烃分子间的作用能,该作用使基础油分子可以吸附在Fe(110)表面,形成稳定的润滑油膜,发挥减摩抗磨作用。
(2)不同结构烃分子之间的作用能不同,其由大到小顺序为:芳香烃,环烷烃,直链烷烃,异构烷烃;不同结构烃分子与金属表面的吸附能也不相同,其由大到小顺序为:芳香烃,直链烷烃,环烷烃,异构烷烃;长直侧链环烷烃是能够保证润滑油膜的稳定性和较低的运动阻力,是理想的基础油组分。
(3)剪切运动不会明显影响润滑油膜的稳定性,并且会使体相中间层分子的极性增加,使分子间的范德华作用增强,从而使分子间作用能增加。
(4)随着温度升高,基础油分子与Fe(110)表面的吸附能及分子间作用能均降低,使润滑油膜的稳定性降低,润滑作用减弱。
(5)不同温度下平衡态时,十四烷基环己烷分子水平分布的吸附能均比竖直分布的吸附能大,说明在平衡润滑状态下,烃分子更容易水平分布在Fe(110)表面。
[1] 刘正林. 摩擦学原理[M].北京: 高等教育出版社, 2009: 62-76.
[2] 张景河. 现代润滑油与燃料添加剂[M].北京: 中国石化出版社, 1991: 160.
[3] 袁晓东, 倪丽, 张建锋, 等. 材料的抗磨减摩机理探讨[J].润滑油, 2011, 26(2): 1-6. (YUAN Xiaodong, NI Li, ZHANG Jianfeng, et al. Discussion on the antiwear and friction-reducing mechanism of materials[J].Lubricating Oil, 2011, 26(2): 1-6.)
[4] 曾新民. 我国润滑油分类及产品标准[J].润滑油, 1992, 7(1): 5-8. (ZENG Xinmin. Lubricating-oil classification and products standard in China[J].Lubricating Oil, 1992, 7(1): 5-8.)
[5] ZHENG R H, LIU G J, DEVLIN M, et al. Friction reduction of lubricant base oil by micelles and crosslinked micelles of block copolymers[J].Tribology Transactions, 2010, 53(1): 97-107.
[6] BART J C J, EMANUELE G, STEFANO C. Lubricants: Properties and characteristics[J].Woodhead Publishing Series in Energy, 2013, 46 (Biolubricants): 24-73.
[7] 李维民, 姜程, 王晓波, 等. 植物油基润滑油添加剂的制备及其摩擦学性能[J].石油学报(石油加工), 2015, 31(2): 468-475. (LI Weimin, JIANG Cheng, WANG Xiaobo, et al. Preparation and tribological of vegetable oil based lubricating oil additive[J].Acta Petrolei Sinica (Petroleum Processing Section), 2015, 31(2): 468-475.)
[8] 吴燕霞, 李维民, 王晓波. 磷系极压抗磨剂在酯类油中的摩擦学性能[J].石油学报(石油加工), 2015, 31(5): 1122-1128. (WU Yanxia, LI Weimin, WANG Xiaobo. Tribological behaviors of P-extreme pressure and anti-wear additives in ester base oil[J].Acta Petrolei Sinica (Petroleum Processing Section), 2015, 31(5): 1122-1128.)
[9] 杨蔚权, 陈波水, 方建华, 等. 油酸甲酯型含氮硼酸酯的合成及其抗磨减摩特性[J].石油学报(石油加工), 2016, 32(1): 82-87. (YANG Weiquan, CHEN Boshui, FANG Jianhua, et al. Synthesis of boric acid ester containing nitrogen from methyl oleate and its tribological performance[J].Acta Petrolei Sinica (Petroleum Processing Section), 2016, 32(1): 82-87.)
[10] JABBARZADEH A, ATKINSON J D, TANNER R I. The effect of branching on slip and rheological properties of lubricants in molecular dynamics simulation of Couette shear flow[J].Tribology International, 2002, (35): 35-46.
[11] LIU P Z, YU H L, REN N, et al. Pressure-viscosity coefficient of hydrocarbon base oil through molecular dynamics simulations[J].Tribology Letters, 2015, 60(3): 1-9.
Molecular Dynamics Simulation on the Lubricating Property of Mineral Base Oil
LI Yiya, LONG Jun, DUAN Qinghua, DAI Zhenyu, ZHAO Yi, SU Shuo
(ResearchInstituteofPetroleumProcessing,SINOPEC,Beijing100083,China)
To study lubricant performance of the base oil molecule, tetradecylcyclohexane was taken as model compound, and the adsorption energy of molecule with Fe(110) surfaces and the intermolecular interaction energy were calculated using molecular dynamics simulation. The influence of molecular structure and temperature on the lubricant performance was also analyzed. The simulation results indicated that the adsorption energy was larger than intermolecular interaction energy by a substantial amount, which could make the lubricant film stable. Moreover, the van der Waals interaction made the greatest contribution to the adsorption energy and intermolecular interaction energy. When the lubricant system reached equilibrium, the horizontal molecule configuration was more stable than the vertical molecule configuration. As of different molecular structures, the decreasing order of their intermolecular interaction energy is: aromatics>cycloalkane>linear alkane>isoalkane, while the decreasing order in terms of their adsorption energy is: aromatics>linear alkane>cycloalkane>isoalkane. Both kinds of energy decreased with the temperature increased, which could result to lower stability of the lubricant film.
base oil; lubricating property; molecular dynamics simulations; adsorption energy; intermolecular interaction energy
2016-08-19
中国石油化工股份有限公司技术开发项目(ST16019)资助
李义雅,女,博士研究生,从事润滑油及分子模拟方面的研究
段庆华,男,教授级高级工程师,从事润滑油方面的研究;Tel:010-82368246;E-mail:duanqh.ripp@sinopec.com
1001-8719(2017)04-0619-07
TE626.3
A
10.3969/j.issn.1001-8719.2017.04.003
