儿童Dent病临床特点及CLCN5基因突变分析
2017-08-09陈国强张小鸽李志娟成银萍
陈国强 张小鸽 李志娟 成银萍
儿童Dent病临床特点及CLCN5基因突变分析
陈国强 张小鸽 李志娟 成银萍
目的 Dent病是一种罕见的X连锁隐性遗传性肾小管疾病,通过探讨儿童Dent病的临床特点和基因特征,旨在提高对儿童Dent病的认识。方法 通过分析3例Dent病患儿的临床资料和CLCN5基因检测结果,复习相关文献,以了解Dent病的表型和基因型,并总结经验。结果 3例患儿的发病年龄1~5岁,确诊年龄1~7岁,首发症状为大量蛋白尿,且证实为低分子蛋白尿,伴有不同程度的高钙尿症,均有镜下血尿。3例患儿的乳酸脱氢酶均正常,均无高血压,无肾功能不全,无贫血,无肾结石或肾钙质沉着症,无肾小管酸中毒,无佝偻病,无矮小症,无智力低下,眼科检查正常,无氨基酸尿及糖尿,无肾脏疾病或尿石症家族史,血清尿素氮、肌酐、白蛋白、钾、钠、氯、钙、镁、磷均正常。基因检测发现3种突变,2例错义突变,1例移码突变,3例患儿均为新生突变;其中c.779G>A突变位点为既往文献报道过的位点,另外发现两个新的突变位点c.1711C>T(E12),c.458(E7)_c.459(E7)insA,既往文献无报道,为新的突变。结论 儿童Dent病临床表现多样,并且临床表现和病程长短有关,通过基因检测可以确诊Dent病。早期发现、早期诊断,可以避免过度免疫抑制及治疗。
Dent病;CLCN5;低分子蛋白尿;高钙尿症
Dent病是一种罕见的X连锁隐性遗传性肾小管疾病。1964年Dent和Friedman首次报道,有2例合并近端肾小管损害、高尿钙和佝偻病表现的英国男孩。1997年发现CLCN5基因的突变可以导致Dent病、X连锁隐性肾石症(XRN;OMIM31-0468)、X连锁隐性遗传性低磷酸盐血症性佝偻病(XLRH;OMIM300554)以及日本儿童特发性低分子蛋白尿(JILMWP;OMIM308990),认为他们是同一种疾病,故将其统称为Dent病。2008年,李鹏等[1]在国内率先建立了基因诊断方法,并首次报道了2例低分子蛋白尿患者,经检测为CLCN5基因突变,确诊Dent病;此后陆续有少量的病例报道,但主要集中在北京、上海、广州等地区[2-7],而西部地区目前尚未有相关报道。随着基因诊断方法引入到西部地区,西安市儿童医院及西北妇女儿童医院肾脏科利用该方法陆续诊断了3例1型Dent病,本研究通过归纳和总结这3例Dent病患儿的临床特点,并对其基因结果进行分析,旨在提高对儿童1型Dent病的认识。
资料和方法
一、研究对象
回顾性分析2014年8月至2016年10月西安市儿童医院及西北妇女儿童医院肾脏科收治的3例Dent病患儿,均为男孩。
二、方法
1.临床资料收集 收集3例患儿入院后的相关资料:一般情况包括姓名、性别、年龄、发病年龄、确诊时间、体质量、身高、血压,并询问家族史;检验项目包括血常规、尿液分析、尿红细胞形态、尿蛋白电泳、尿α1和β2微球蛋白、微量白蛋白、转铁蛋白、免疫球蛋白、24h尿蛋白定量、24h尿钙定量、尿钙尿肌酐比值测定、肝功能、肾功能、心肌酶、电解质、血气分析等;检查项目包括肾脏B超、眼科检查、骨骼X线片、智力测评。经患儿父母同意并签署知情同意书后,分别采取患儿及其父母空腹血样5 ml,采用盐析法提取基因组DNA,通过检索数据库及相关文献,设计肾病相关基因二代测序靶向捕获试剂盒,以上监测由康旭医学检验所完成。
2.相关诊断标准 1型Dent病诊断标准符合以下4条[8]:①小分子量蛋白尿,即尿ɑ1微球蛋白明显升高或尿蛋白电泳以小分子量蛋白为主;②高钙尿症,即尿钙/肌酐比大于0.25或24h尿钙大于4 mg/kg;③至少有下列情况之一,肾钙化、肾石症、血尿、低磷血症、肾功能不全;④其突变基因为CLCN5基因。本组3例患儿均符合以上1型Dent病诊断标准。
结 果
一、临床表现及实验室检查结果
3例患儿的发病年龄1~5岁,确诊年龄1~7岁,首发症状为大量蛋白尿,且证实为低分子蛋白尿,伴有不同程度的高钙尿症,且均有镜下血尿。3例患儿的乳酸脱氢酶正常,无高血压,无肾功能不全,无贫血,无肾结石或肾钙质沉着症,无肾小管酸中毒,无佝偻病,无矮小症,无智力低下,眼科检查正常,血清尿素氮、肌酐、白蛋白、钾、钠、氯、钙、镁、磷均正常,无氨基酸尿及糖尿,无肾脏疾病或尿石症家族史。(表1)

表1 3例患儿主要实验室检查结果
二、基因检测结果
3例患儿基因检测均发现CLCN5基因突变。例1为c.779G>A,该基因的编码区779位的碱基G突变为A;该变异导致从第260号氨基酸蛋白位置上,氨基酸G突变为氨基酸D(p.G260D),为错义突变。其母该位点未见异常,其父未检测,该患儿为新生突变。例2为c.1711C>T(E12),该基因的编码区1711位的碱基C突变为T;该变异导致第571氨基酸蛋白位置上,氨基酸L突变为氨基酸F(p.L571F),为错义突变。其父该位点未见异常,其母该位点为杂合子。该患儿基因突变来自母亲遗传。该基因突变目前未见报道,应为新的突变。例3为c.458(E7)_c.459(E7)insA,该基因的编码区458至459位中间插入碱基A;该变异导致从第153号氨基酸开始,蛋白再往下翻译15个氨基酸后终止翻译(p.T153fs*15),为移码变异。其父母亲该位点均未见异常,该患儿为新生突变。(表2)
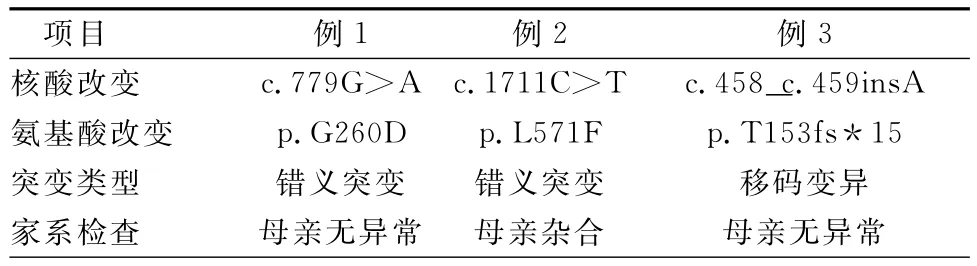
表2 3例患儿CLCN5基因突变结果
讨 论
1997年研究发现CLCN5基因的突变可以导致Dent病,2004年Hoopes等[9]报道OCRL1基因突变也可引起Dent病表现。因此,目前根据突变基因的不同将Dent病分为两型,突变基因位于电压门控性氯离子通道蛋白5(Chloride Voltage-Gated Channel 5,CLCN5)基因上的为1型Dent病,突变基因位于眼-脑-肾综合征1(oculocerebrorenal syndrome of Lowe-1,OCRL-1)基因上的为2型Dent病[10]。绝大部分的报道均为单基因致病,Addis等[11]报道了1例由CLCN5与OCRL-1基因共同突变引起的儿童病例。
Dent病临床特征主要是低分子蛋白尿、高钙尿、肾钙质沉着以及进行性肾衰竭,其中30%~80%患者在30~50岁时发展至肾病终末阶段[12],但日本等报道的肾衰竭发生率仅8%左右[13]。其他临床表现主要包括氨基酸尿(40%~50%),磷酸盐流失症(20%~25%),钾流失症(5%~15%),糖尿(10%~20%),肾小管酸中毒(3%~5%)以及成年后肾衰竭。尽管肾钙质沉着症和高钙尿是Dent病的特征表现,但所有患者均出现了低分子量蛋白尿[14-15]。目前发现Dent病患者表型谱在扩大,有一部分患者只表现为低分子量蛋白尿,而尿钙水平完全正常[16];而另一部分患者却表现为肾病范围蛋白尿,尿蛋白电泳为非选择性蛋白尿[17]。Blanchard等[18]回顾分析了109例CLCN5基因突变(Dent病1型)以及9例OCRL突变(Dent病2型)病例,发现在Dent病1型中,随着年龄的增长,估算肾小球滤过率下降,有或无肾钙质沉着症患者之间无差异;低分子蛋白尿的中位数在肾病范围,即使在晚期肾脏疾病也保持在同一水平;12例患者发生终末期肾病,平均年龄40岁;随着年龄增长,低磷酸盐血症持续存在,骨化三醇浓度维持在正常范围;并且随着年龄的增长,血钾降低,有一半的患者18岁以后出现低血钾。本研究这3例患儿均表现为低分子蛋白尿和高钙尿症以及镜下血尿,无肾功能不全,无肾石症或肾钙质沉着症,无肾小管酸中毒,无佝偻病,无低钾低磷酸血症等其他表现,结合文献报道分析,3例患儿中年龄最大者7岁,最小者1岁,病程最长者仅2年,考虑由于患者年龄小,病程短,很多症状没有表现出来,推测随着年龄的增长会有其他表型出现。对比国内的其他患者,Li等[7]的报道中,4/15患者表现出明显的电解质紊乱和酸中毒的表现,但这4例患者病程在7~16年;1/15患者有肾石症,8/15患者有佝偻病的表现。简珊等[3]报道中,1/4患者有肾石症,2/4患者有肾钙质沉着症,3/4患者有低磷血症,3/4患者有佝偻病,病程在间4.5~7.8年。因此,Dent病的临床表现的多少和病程长短有一定的联系,我们还需要更多的病例以及更长的随诊时间来观察西部地区患儿的表型特点。
基因突变是Dent病的发病原因,目前所检测到的突变中,CLCN5基因突变占60%,OCRL基因突变占15%[19]。CLCN5基因位于染色体Xp11.22-11.23,编码746个氨基酸长度的CLC-5蛋白。CLC-5蛋白属于电压门控性氯离子通道蛋白家族成员,具有高度保守的结构,在体内发挥着重要作用,包括调节膜的兴奋性、维持细胞内外离子的稳态和调节内吞体酸化功能。由CLCN5基因突变所引起的1型Dent病患者主要临床表现包括肾小管功能障碍、LWMP、高钙尿、肾脏钙化及肾结石,部分患者出现进行性肾衰竭。在肾小管上皮细胞,早期内吞体负责重吸收由肾小球滤过的低分子蛋白。正常情况下,内吞体酸化引起Megalin/Cubilin多配体复合体解离,同时Megalin/Cubilin在顶膜回收再利用,而其余配基被溶酶体直接降解。CLCN5基因突变引起的氯离子内流受限、内吞体酸化异常可导致Megalin/Cubilin复合体解离异常,令受体介导的胞吞作用受损,出现维生素D结合蛋白、25羟维生素D3等小分子蛋白尿丢失增加[20-21]。
迄今为止,全世界范围报道了大约有250个Dent病Ⅰ型家系,致病突变分散在整个基因谱,突变谱包括错义突变(44%),无义突变(26%),小缺失/插入(15%),大缺失(4%),剪切点突变(11%),伴有几个热点,主要影响精氨酸密码子[19]。我们这3例患者,2/3为错义突变,1/3为移码变异。其中例1患者260位氨基酸G突变为氨基酸D(p.G260D),该突变为已经报道过的突变[22],母亲该位点无异常,提示该患儿为新生突变;例2患者571位氨基酸L突变为氨基酸F(p.L571F),目前未见报道,应为新的突变,其父该位点未见异常,其母该位点为杂合子,该患儿基因突变来自母亲遗传。这两例患者均为错义突变,其他位氨基酸蛋白无改变,因此推测患儿临床症状轻,预后相对较好。例3患者其父母亲该位点均未见异常,该患儿为新生突变。基因编码区458至459位中间插入碱基A,目前未见报道,应新的突变,该移码变异使蛋白翻译提前终止,造成基因编码蛋白截短。因此推测该患儿临床症状重,预后相对较差。
儿童Dent病临床表现多样,并且临床表现和病程长短有关,目前关于Dent病的表型谱和基因谱还在不断有新的报道出现,对于蛋白尿的患儿,无论是否属于肾病范围蛋白尿,都应该行尿蛋白成分的检测,如果为低分子蛋白尿,无论是否合并高钙尿症均应注意Dent病的可能,尽早行基因检测以明确诊断,避免不必要的免疫抑制剂治疗。
[1] 李鹏,黄建萍.小儿Dent病临床特征和基因突变分析[J].临床儿科杂,2008,26(4):298-301.
[2] 朱碧溱,李鹏,黄建萍.以小分子蛋白尿为主要表现的六例临床及基因分析[J].中华儿科杂志,2010,48(5):329-332.
[3] 简珊,魏珉,何艳燕,等.Dent病4例临床及基因分析[J].中国当代儿科杂志,2015,17(12):1261-1266.
[4] Ji LN,Chen CY,Wang JJ,et al.A novel CLCN5 mutation in a Chinese boy with Dent's disease[J].World J Pediatr,2014,10(3):275-277.
[5] 张宏文,张琰琴,刘晓宇,等.Dent病6例临床诊治分析[J].临床儿科杂志,2016,34(6):418-420.
[6] 李国民,方晓燕,徐虹,等.儿童2型Dent病1例并文献复习[J].中国循证儿科杂志,2014,9(6):456-459.
[7] Li F,Yue Z,Xu T,et al.Dent Disease in Chinese Children and Findings from Heterozygous Mothers:Phenotypic Heterogeneity,Fetal Growth,and 10 Novel Mutations[J].J Pediatr,2016,174:204-210.
[8] Hoopes RR Jr,Raja KM,Koich A,et al.Evidence for genetic heterogeneity in Dent'S disease[J].Kidney Int,2004,65(5):1615-1620.
[9] Hoopes RR Jr,Shrimpton AE,Knohl SJ,et al.Dent disease with mutations in OCRL1[J].Am J Hum Genet,2005,76(2):260-267.
[10]Devuyst O,Thakker RV.Dent's disease[J].Orphanet J Rare Dis,2010,5:28.
[11]Addis M,Meloni C,Tosetto E,et al.An atypical Dent's disease phenotype caused by co-inheritance of mutations at CLCN5 and OCRL genes[J].Eur J Hum Genet,2013,21(6):687-690.
[12]Tosetto E,Ghiggeri GM,Emma F,et al.Phenotypic and genetic heterogeneity in Dent's disease-the results of an Italian collaborative study[J].Nephrol Dial Transplant,2006,21(9):2452-2463.
[13]Sekine T,Komoda F,Miura K,et al.Japanese Dent disease has a wider clinical spectrum than Dent disease in Europe/USA:genetic and clinical studies of 86 unrelated patients with lowmol ecular-weight proteinuria[J].Nephrol Dial Transplant,2014,29(2):376-384.
[14]B kenkamp A1,B ckenhauer D,Cheong HI,et al.Dent-2 disease:a mild variant of Lowe syndrome[J].J Pediatr,2009,155(1):94-99.
[15]Ludwig M,Utsch B,Balluch B,et al.Hypercalciuria in patients with CLCN5 mutations[J].Pediatr Nephrol,2006,21(9):1241-1250.
[16]De Mutiis C,Pasini A,La Scola C,et al.Nephrotic-range Albuminuria as the presenting symptom of Dent-2 disease[J].I-talian Journal of Pediatrics,2015,41:46.
[17]Cramer MT,Charlton JR,Fogo AB,et al.Expanding the phenotype of proteinuria in Dent disease[J].A case series.Pediatr Nephrol,2014,29(10):2051-2054.
[18]Blanchard A,Curis E,Guyon-Roger T,et al.Observations of a large Dent disease cohort[J].Kidney Int,2016,90(2):430-439.
[19]Ludwig M,Levtchenko E,B kenkamp A.Clinical utility gene card for:Dent disease(Dent-1 and Dent-2)[J].Eur J Hum Genet,2014,22(11).DOI:10.1038/ejhg.2014.33.
[20]Saito A,Sato H,Iino N,Takeda T.Molecular mechanisms of receptor-mediated endocytosis in the renal proximal tubular epithelium[J].J Biomed Biotechnol,2010:403272.
[21]Christensen EI,Devuyst O,Dom G,et al.Loss of chloride channel ClC-5 impairs endocytosis by defective trafficking of megalin and cubilin in kidney proximal tubules[J].Proc Natl Acad Sci U S A,2003,100(14):8472-8477.
[22]Tosetto E,Graziotto R,Artifoni L,et al.Dent's disease and prevalence of renal stones in dialysis patients in Northeastern Italy[J].J Hum Genet,2006,51(1):25-30.
Clinical and CLCN5 genetic mutation analysis of Dent's disease in children
CHEN Guo-qiang,ZHANG Xiao-ge,LI Zhi-juan,CHENG Yin-ping.
Department of Nephrology,Northwest Women's and Children's Hospital,Xi'an 710061,China
Corresponding author:ZHANG Xiao-ge,E-mail:zxge007@163.com
Objective Dent disease is a rare X-linked recessive renal tubular disease.This study aimed to enhance the recognition of dent disease by exploring the clinical characteristics and genetic features.Methods Methods The clinical data of 3 children with Dent disease,genetic test results for CLCN5,and relevant literatures were analyzed retrospectively for further recognizing the clinical/genetic phenotype of Dent disease and subsequently summarizing experience.Results The onset age of 3 children was 1-5 years,while the age at diagnosis was 1-7 years.Massive proteinuria,which was subsequently proved to be low molecular weight proteinuria(LMWP),was defined as the initial symptom in all patients.Meanwhile,varying degrees of hypercalciuria and microhematuria were also observed.All 3 patients displayed normal activity of lactate dehydrogena,no hypertension,no renal insufficiency,no anemia,no renal calculus or nephrocalcinosis,no renal tubular acidosis,no rickets or dwarfism,no mental retardation,normal ophthalmic examination,no aminoaciduria or glycosuria,no family history of kidney diseases,and normal level of serum urea nitrogen,creatinine,albumin,potassium,sodium,chlorine,calcium,magnesium and phosphate.Genetic testing demonstrated 3 de novo mutations:2 of missense mutation and 1 of phase-shift mutation.Among them,the mutation of c.779G>A had been reported in previous studies,while others of c.1711C>T(E12)and c.458(E7)_c.459(E7)insA were the first discovered mutations.Conclusions The clinical manifestations of Dent disease,which are also related to the disease course,are varied among children.Early detection and diagnosis of Dent disease by genetic testing may be an effective strategy to avoid excessive immunosuppressive-therapy.
Dent disease;CLCN5;Low molecular weight proteinuria;Hypercalciuria
2016-10-17
2017-04-15)
10.3969/j.issn.1671-2390.2017.07.005
710061 西安,西北妇女儿童医院肾内科(陈国强,张小鸽);710003 西安,西安市儿童医院肾内科(陈国强,张小鸽,李志娟,成银萍)
张小鸽,E-mail:zxge007@163.com
