阴阳离子聚氨酯/羟基磷灰石复合微球的制备及其自组装
2017-08-07李玉宝
赖 欣 侯 毅 李玉宝 黄 敏 任 欣 张 利
阴阳离子聚氨酯/羟基磷灰石复合微球的制备及其自组装
赖 欣 侯 毅 李玉宝 黄 敏 任 欣 张 利*
(四川大学纳米生物材料研究中心,分析测试中心,成都 610064)
分别以2,2-双羟基丙酸(DMPA)和N-甲基二乙醇胺(MDEA)作扩链剂,采用自乳化法成功制备出带有正、负电荷的阳、阴离子聚氨酯/羟基磷灰石(PU/HA)微球,并考察HA的复合比例对微球形貌、理化性能的影响。在此基础上,通过微球间的静电作用力,将制备出的阴、阳离子复合微球自组装形成凝胶体系,并对其力学性能进行探究。SEM、ζ电位和激光粒度结果显示成功制备出具有一定形貌的带电微球,其中当扩链剂含量为15%(n/n)、HA含量为10%(w/w)时,微球形貌较佳且粒度分布均匀;X射线衍射、热分析结果表明,微球确为一定比例的HA与PU复合产物;自组装体系的流变学测试结果表明,微球组装后的产物呈类凝胶状态,具有较高的弹性模量,并有一定的可注射性。同时,骨髓间充质干细胞(BMSCs)可在组装材料表面粘附、铺展,生长良好。
阴离子PU/HA微球;阳离子PU/HA微球;自乳化;自组装;可注射凝胶
近年来,微创治疗因其创口小、并发症少、疗效确切等优点在临床治疗上得到广泛应用[1-2]。可注射性凝胶作为微创治疗的一类重要材料,自然成为生物医学材料领域的一大研究热点[3-9]。这类材料具有生物相容性好、形状可塑、理化性能可调控等优点,但其功能相对单一、稳定性相对较弱等缺陷严重限制了其发展。近年来,有研究者[10-19]以微/纳米球材料为基本结构单元,利用微球间的静电力、磁场力和空间位阻等作用力诱导自组装构建微球基可注射水凝胶材料,如Wang等[10]设计制备出分别带正/负电荷的微米级或纳米级高分子微球,并利用微球间静电作用力驱使带相反电荷的微/纳米微球,通过“自下而上”的制备方式自组装构建出更大尺寸的稳定凝胶态材料;Cheng等[11]利用聚合物微球链之间的相互作用,也是通过“自下而上”的制备方式自组装构造3D多孔支架。与其他凝胶体系相比,微球基自组装凝胶体系表现出较高的力学强度,优良的可注射性和可塑性。此外,通过物理作用力组装形成的微球基凝胶体系可避免化学交联过程中有毒副作用的化学物质引入,更适于生物医学方面的应用,有望弥补目前凝胶材料的不足。
聚氨酯(Polyurethane,PU)因其良好的生物相容性、机械性能和易加工性,在医学材料领域有着广泛的应用[20-23]。其分子主链是由玻璃化温度低于室温的软段和玻璃化温度高于室温的硬段嵌段组成,在聚氨酯材料的制备过程中,可通过调节软硬段的成分、比例,使其具有不同化学结构、力学性能等理化性质[24-26],以满足不同的应用需求。通过自乳化法制备PU荷电微球,可在制备过程中引入含阴、阳离子基团的亲水链端,使带有相反电荷的PU微球在静电引力作用下实现自组装。
羟基磷灰石(Hydroxyapatite,HA)是一类化学成分和结构与生物体骨组织相似,具有良好生物活性和生物相容性的材料[27],且无毒、无免疫原性,具有良好的骨传导能力,因此在骨修复领域已有着广泛的应用。但HA在单独使用时存在脆性大、可塑性差、易团聚等缺点,为改善其使用性能,研究者们通常将HA与高分子基体复合,制备兼有HA优良生物相容性和高分子材料良好力学性能的有机/无机复合材料[28-31]。
本文分别以2,2-双羟基丙酸(DMPA)和N-甲基二乙醇胺(MDEA)作为扩链剂,采用自乳化法制备阴、阳离子PU微球,在此基础上复合一定量的HA,在改善材料生物相容相性、生物活性的同时提高材料的力学性能。通过对制备工艺条件的调节,探究扩链剂、HA对PU、PU/HA微球成球性、表面形态及理化性能等的影响,寻求最佳合成条件。最后在静电力的作用下,将制得的阴、阳离子PU/HA复合微球自组装成凝胶体系,并对其力学性能及可注射性进行分析、探究,为微球基可注射凝胶材料的研究奠定一定的理论基础。
1 实验部分
1.1 实验原料
异氟尔酮二异氰酸酯 (IPDI),聚四氢呋喃(PTMG-2000),异辛酸亚锡,N,N-二甲基甲酰胺(DMF),2,2-双羟基丙酸 (DMPA),N-甲基二乙醇胺(MDEA),冰乙酸,丙酮均来自Aladdin试剂公司。三乙胺,无水乙醇,四水合硝酸钙(Ca(NO3)2·4H2O),十二水合磷酸三钠(Na3PO4·12H2O)均来自成都科龙化工试剂厂。去离子水,实验室自制。
在聚氨酯聚合反应中,异氰酸酯会与原料中含有的水分发生副反应,为避免这一情况发生,在实验前需对部分原料进行脱水处理。PTMG-2000在120℃烘箱中干燥2 h,丙酮、TEA用3A分子筛提前一周处理。
1.2 阴、阳离子PU微球的制备
将26.67 g IPDI,24 g PTMG-2000加入到 100 mL三口烧瓶中,置于82℃的油浴锅中,加入2滴异辛酸亚锡,低速搅拌,进行预聚反应。3 h后,降温至52℃,将溶有物质的量分数为5%、10%、15%的DMPA/MDEA的DMF(8 mL)溶液加入到反应体系中,进行扩链反应。上述过程中,根据反应体系的粘度情况,加入适量的丙酮(约10~30 mL),用以降低反应体系粘度。反应进行2 h后,分别将0.71、1.52、2.33 g三乙胺/0.42、0.90、1.38 g乙酸分散到 400 g去离子水中配制中和液,室温高速搅拌下缓慢将聚氨酯预聚物滴入中和液中进行中和反应,剪切乳化2 h得PU微球。将得到的产物多次清洗、离心、冻干,以备后用。
1.3 载HA阴阳离子聚氨酯微球的制备
1.3.1 HA的制备
称取114.06 g Na3PO4·12H2O溶于1 000 mL去离子水中,称取 118.08 g Ca(NO3)2·4H2O溶解于1 000 mL去离子水中,完全溶解后,将硝酸钙溶液置于70℃水浴条件下,通过分液漏斗,将磷酸三钠溶液逐滴滴入硝酸钙溶液中,并加入聚乙二醇作为分散剂,约1 h滴完,同时以1 000 r·min-1的转速进行搅拌。滴完后继续保持搅拌20 min,并在此期间滴加4 mol·L-1的NaOH溶液,调节其pH值至9~10。洗涤至pH值为中性,冻干后,留做备用。
1.3.2 载HA阴、阳离子PU微球的制备
将26.67 g IPDI,24 g PTMG-2000加入到100 mL三口烧瓶中,置于82℃的油浴锅中,加入2滴异辛酸亚锡,低速搅拌,进行预聚反应。3 h后,降温到52℃,分别将溶有物质的量分数为5%、10%、15%的DMPA/MDEA的DMF(8 mL)溶液加入到反应体系中,进行扩链反应,同时将一定比例的HA粉末(HA的质量分数分别为5%、10%、15%)加入预聚体中,进行充分的搅拌混合。根据反应体系的粘度情况,加入适量的丙酮(约10~30 mL)用以降低反应体系粘度。反应进行2 h后,分别将0.71、1.52、2.33 g三乙胺/0.42、0.90、1.38 g乙酸分散到400 g去离子水中配制中和液,中和液与扩链后的聚合物进行中和反应,剧烈搅拌2 h。将得到的产物多次清洗、离心、冻干,以备后用。
1.4 阴阳离子聚氨酯微球的自组装
将上述制得的HA质量分数为0%、5%、10%、15%的阴、阳离子微球(扩链剂含量为15%(n/n))分别分散于去离子水中(固含量为0.15 g·mL-1),数小时后将相同HA含量的阴、阳离子微球悬浊液混合使其进行自组装。
1.5 细胞培养实验
将4周龄的SD大鼠断颈处死后迅速在无菌条件下取出双侧股骨和胫骨。用10 mL无菌注射器吸取α-MEM培养基冲出骨髓,所获细胞用5 mLα-MEM培养基(含双抗)制成细胞悬液。将细胞悬液直接种入玻璃培养瓶内,并于37℃、5%(V/V)CO2的培养箱内培养,24 h半换液,48 h全换液。待贴壁细胞融合到90%时,按1∶3比例传代。取第3代骨髓间充质干细胞(BMSCs)用于与材料复合培养实验。
将纯PU、PU/HA(HA质量分数10%)组装材料在无菌条件下分别置于24孔板中,将上述传代培养的BMSCs按2×104/孔的浓度滴加在孔板中,每孔滴加1 mL,待细胞自然沉降在材料表面后,又于37℃的5%CO2细胞培养箱中培养,12、48 h取样。样品取出后用PBS冲洗,去除未贴壁细胞,经3%戊二醛固定2 h,50%~100%乙醇梯度脱水,临界点干燥,干燥镀金后用扫描电镜观察。
1.6 表征方法
本文采用扫描电子显微镜 (JSM-6510LV,日本JEOL公司)观察微球形貌尺寸和分散性,以及细胞与组装材料共培养后的生长形貌;样品平均粒径采用激光粒度仪(2000E,丹东百特仪器有限公司)进行测试分析;样品的表面电荷通过ζ电位测试仪(ZS90型,英国马尔文仪器有限公司)测试;用X射线衍射仪(DX-2500,丹东通达科技有限公司)表征样品的物相成分,Cu Kα,λ=0.154 06 nm,U=40 kV,I=25 mA,2θ= 5°~80°;用热分析仪(STA 499,德国耐驰公司)对样品的热稳定性进行分析,升温范围为25~500℃,升温速率10℃·min-1;使用流变仪(DHR-2,美国TA公司)对组装体材料的力学性能及可注射性进行测评。
2 结果与讨论
2.1 PU微球和PU/HA复合微球的SEM照片
采用SEM对不同扩链剂含量的阴、阳离子PU微球形貌进行观察,如图1所示。从图1a和图1d可以看出,当扩链剂含量为5%(n/n)时,制备产物粘连在一起,无明显形状。当扩链剂含量为10%(n/n) (图1b和图1e)时,制备产物大多呈球状,但微球粒径分布相对不均,且微球表面粗糙。随着扩链剂含量的不断增加,当扩链剂含量为15%(n/n)(图1c和图1f)时,制备产物球形度相对较好,粒径分布均匀,且表面光滑。这可能是由于随着扩链剂含量增加,亲水基团增多,使得扩链后的产物亲水性增强,易于在水溶液中乳化成球;水分子进入聚氨酯分子链间的速度加快,乳化形成微球的过程所需时间减少,因此每个微球形成的时间间隔就越小,微球也就越均匀[32]。

图1 不同扩链剂含量的阴、阳离子PU微球的SEM图Fig.1 SEM images of anionic(a~c)and cationic(d~f)PU microspheres with different chain extenders contents(n/n)of(a,d)5%,(b,e)10%,(c,f)15%
基于上述实验,优化扩链剂含量为15%(n/n)的微球制备工艺参数,制得PU/HA复合微球。SEM照片可以看出,当HA质量分数为5%(图2a,图2d)时,微球表面相对粗糙,有少量HA附着于微球表面;同时,由于大部分HA与PU混合,占据了微球内部一定空间,使得微球粒径相对增大。随着HA含量增加,微球表面有更多HA颗粒暴露(图2b,图2e),可显著提高材料的生物活性;而当HA质量分数达15%时,如图2c、2f所示,产物球形度相对较差,出现粘连现象,可能是随着HA含量不断增加,大量HA粉末团聚,未能与PU预聚体均匀混合,影响了聚合物的乳化,进而影响了产物形态。

图2 不同HA含量阴、阳离子PU/HA微球的SEM图Fig.2 SEM images ofanionic(a~c)and cationic(d~f) PU/HA microspheres(15%(n/n)chain extenders contents)with HA contents(w/w)of(a,d)5%; (b,e)10%;(c,f)15%
2.2 平均粒径及ζ电位测试
HA含量不同的阴、阳离子PU/HA微球的激光粒度测试、ζ电位测试结果如表1、2所示。
表1 不同HA含量的阴、阳离子PU/HA微球平均粒径
Table 1 Average particle size of oppositely charged PU/HA microspheres

Mass fractin of HA/%(w/w) Average particle size/μm Anionic PU microspheres Cationic PU microspheres 0 10.72±0.90 10.76±0.88 5 12.17±0.54 12.69±0.72 10 16.80±0.24 16.37±1.15 15 11.69±0.57 12.87±0.70
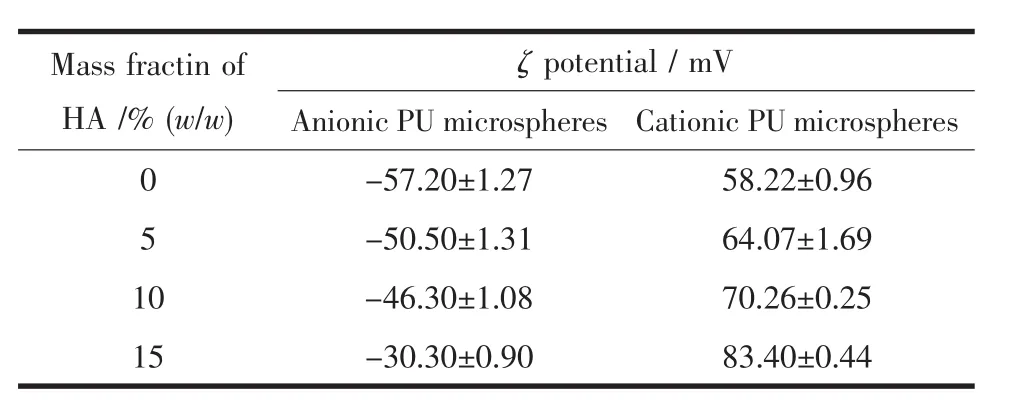
表2 不同HA含量的阴、阳离子PU/HA微球的ζ电位Table 2 ζpotential of oppositely charged PU/HA microspheres
由表1可以看出,随着HA含量增加(0%~10% (w/w)),复合微球粒径逐渐增大,大部分HA粉末包裹于微球内部,仅少量附着于微球表面。而当HA的含量增加到15%(w/w)时,微球粒径减小,这是由于反应中过多的HA粉末未能很好地分散在反应体系中,易于发生团聚,且影响了整个体系的粘度,影响了乳化过程中微球的形成。该测试结果也验证了上述SEM观察的结果。
由表2看出,随着HA含量增加,阴离子微球的ζ电位绝对值逐渐减小,而阳离子微球的ζ电位绝对值逐渐增大,这是由于附着在微球表面的HA本身带有一定的正电,进一步影响了阴、阳离子PU微球的带电情况。当HA质量分数增加到15%时,由于大量HA暴露,附着于微球的外部,所以导致样品带电量变化较大。
2.3 XRD衍射分析
不同HA含量阴、阳离子PU/HA微球的XRD图如图3A、B所示。其中,a、b、c分别表示HA质量分数为5%,10%,15%的PU/HA复合微球。
图中2θ为20°处的强衍射峰是聚氨酯的结晶衍射峰。从图中a、b、c可以看出,2θ为26°和34°处出现了HA晶体(002)和(211)晶面的特征衍射峰,在40°、47°和50°处还可以观察到HA的其他特征衍射峰,与标准卡片JCPDS 09-0432相吻合。随着HA含量增加,衍射峰逐渐增强,说明HA和聚氨酯微球是物理结合。

图3 不同HA含量的阴离子(A)、阳离子(B)PU/HA微球的XRD图Fig.3 XRD patterns of anionic(A)and cationic(B)PU/HA microspheres with HA contents(w/w)of(a)5%,(b)10%,(c)15%
2.4 热重分析
从图4中可以看出,PU/HA微球的分解主要分为两个阶段,分别对应着聚氨酯中硬段和软段的分解。在25~250℃范围内,分解速度较为缓慢,主要是聚氨酯分子链中的氨基甲酸酯分解为异氰酸酯和多元醇,进而分解成二氧化碳等物质;当温度升至250℃后,多元醇开始分解,释放出小分子的物质。460℃以后PU完全分解,由于HA的分解温度为1 330℃,因此550℃时剩下还未分解的HA。如图4A、B中曲线a、b、c所示,反应结束后剩余物质的质量分数分别为5.95%、11.55%、16.34%(图4A),以及5.10%、11.27%、14.81%(图4B),与实验设计的5%、10%、15%有较小偏差,可能是由实验过程中少量杂质引入或其他操作误差引起。
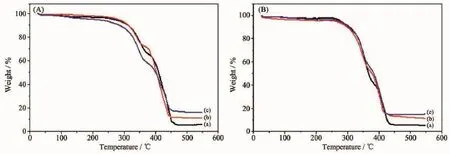
图4 不同HA含量的阴离子(A)、阳离子(B)PU/HA微球的TG曲线Fig.4 TG curves ofanionic(A)and cationic(B)PU/HA microspheres with HA contents(w/w)of(a)5%,(b)10%,(c)15%
2.5 自组装凝胶体系的流变学性质分析
将相同HA含量,固含量为0.15 g·mL-1的阴、阳离子PU/HA微球分别组装在一起,自组装产物的挤出状态照片如图5所示。
从图中可以看出,HA质量分数为0%,5%,10%的3组材料基本组装成型,且挤出物具有一定的形状和粘弹性,这是由于组成组装体的阴、阳离子PU/ HA微球的电位绝对值相对较高,且电量绝对值相差较小,因此带电微球在较强的静电力作用下组装良好。而HA质量分数为15%的阴、阳离子PU/HA微球组装体难以成形,呈现流体状态,这可能由于PU聚合体的包覆能力有限,且较大含量的HA易于团聚,因此HA未能与PU均匀混合,难以成球,进而影响了微球表面的带电情况,使得阴、阳离子微球的电位绝对值相差较大,难以匹配。
如图6所示,将HA质量分数为10%、固含量为0.15 g·mL-1的阴、阳离子PU/HA微球溶液和带相反电荷微球的混合物加入离心管倒置1 min后,阴、阳离子微球溶液仍具有很强的流动性,而对应相同固体含量的带相反电荷PU/HA微球混合物组装沉积滞留于原位,说明其形成了稳定的凝胶。为观察凝胶的微观形貌,对其进行扫描电镜观察,如图7所示,从图中可看出微球之间相互堆积连接,形成三维多孔结构,进一步证实凝胶是通过微球间的静电作用力自组装形成。
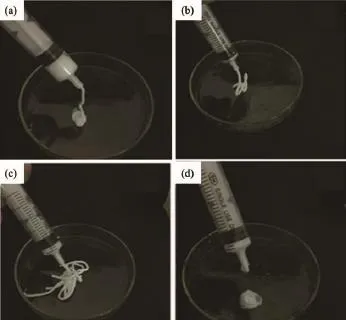
图5 载不同HA含量的阴阳离子PU/HA微球自组装产物挤出状态Fig.5 Extrusion state of gel system self-assembled by oppositely charged PU/HA microspheres with HA contents(w/w)of(a)0%,(b)5%,(c)10%, (d)15%

图6 HA质量分数为10%、固含量为0.15 g·mL-1的阴、阳离子PU/HA微球溶液和带相反电荷微球混合物的样品静置(A)和倒置(B)后的照片Fig.6 Photographs of anionic and cationic PU/HA microspheres(10%(w/w)HA content,15% solid content)and mixtures ofoppositely charged microspheres before(A)and after(B) an inverted-vialtest

图7 HA质量分数为10%、固含量为0.15 g·mL-1的阴、阳离子PU/HA微球自组装形成凝胶的SEM图像Fig.7 SEM images of gel system self-assembled byoppositely charged PU/HA microspheres(10% (w/w)HA content,0.15 g·mL-1solid content)
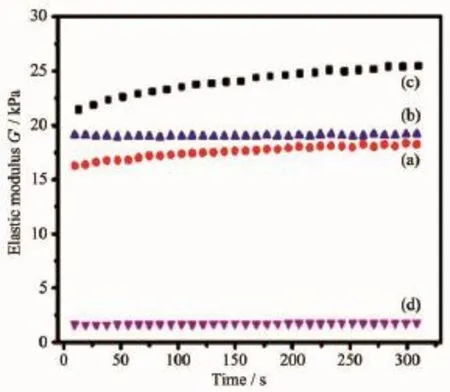
图8 不同HA含量的阴、阳离子PU/HA微球自组装产物的弹性模量Fig.8 Elastic modulus of gel system self-assembled by oppositely charged PU/HA microspheres with HA contents(w/w)of(a)0%,(b)5%,(c)10%,(d) 15%
使用流变仪对组装材料的弹性模量、粘弹性、剪切降粘性质进行测试,结果如图8、9、10所示。由图8可看出,当HA质量分数为0%、5%和10%时,组装材料的弹性模量逐渐增大,当HA质量分数达到15%时,其弹性模量显著降低。说明加了适量HA (5%或10%(w/w))的组装体材料表现出较好的粘弹性,这是由于加入的HA起到了增强作用,复合材料的弹性相应增强;当加入更多HA(15%(w/w))时,会导致成球效果差,在自组装过程中,因微球成形相对较差、大量HA散落且表面所带电荷绝对值相差较大,阴阳离子微球难以组装成形,材料表现出明显的流动性,但却牺牲了较多的弹性模量。从图9中可以发现4组自组装产物的tanδ值均小于1,暗示在HA质量分数为0~15%范围内,阴、阳离子PU/HA微球自组装产物表现出弹性模量大于粘性模量。
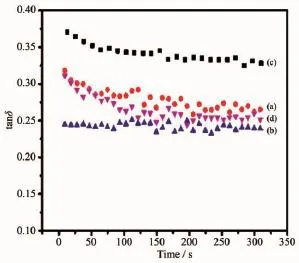
图9 不同HA含量的阴阳离子PU/HA微球自组装产物的粘弹性Fig.9 Viscoelasticity of gel system self-assembled by oppositely charged PU/HA microspheres with HA contents(w/w)of(a)0%,(b)5%,(c)10%, (d)15%
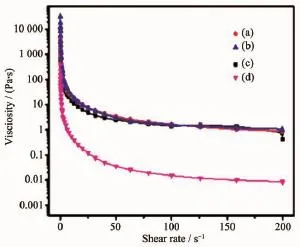
图10 不同HA含量的阴阳离子微球自组装产物的剪切降粘行为Fig.10 Shear bonding behavior of gel system selfassembled by oppositely charged PU/HA microspheres with HA contents(w/w)of (a)0%,(b)5%,(c)10%,(d)15%
图10 是HA质量分数为0~15%的阴阳离子微球基凝胶粘度随剪切速率变化的关系。HA质量分数为0~10%的微球基凝胶在低的剪切速率下具有高粘度,且试验中组装产物都表现出剪切变稀行为(Shear-thinning behavior)[33],即样品的粘度随着剪切速率的增加而减弱,这是因为在高剪切速率下,原本与周围颗粒形成能量壁垒的纳米微球在不断加速的剪切力下发生位移,使得物理交联的网络无法瞬间重建,因而造成了材料表现出低粘度,这与Wang等报道的PLGA纳米微球体系表现出相似的特性[34]。
2.6 细胞相容性
将骨髓间充质干细胞(BMSCs)与阴、阳离子纯PU(扩链剂物质的量分数15%)、PU/HA微球(扩链剂物质的量分数15%,HA质量分数10%)组装材料共培养12 h、48 h后,通过扫描电镜(SEM)观察细胞生长状况,如图11所示。与材料共培养12 h后,材料表面均附着有细胞,纯PU微球组装材料表面粘附的细胞呈梭形,PU/HA组装材料表面细胞呈多边形伸展粘附在材料表面,且有伪足伸出;48 h后,两组材料表面均附着有更多的细胞,与纯PU组装材料相比,PU/HA组装材料表面的细胞铺展状态更好,组装材料上细胞间伪足相连,形成单细胞层。这说明PU/HA组装材料具有更好的细胞相容性,有利于成骨细胞的增殖,有望在骨缺损治疗领域得以应用。

图11 骨髓间充干细胞(BMSCs)与纯PU(a,b)或PU/ HA(c,d)组装材料共培养 (a,c)12 h、(b,d)48 h后的SEM图像Fig.11 SEM images of BMSCs co-cultured with PU(a,b) or PU/HA(c,d)self-assembled material for(a,c) 12 h and(b,d)48 h
3 结 论
本实验采用自乳化法成功制备出阴、阳离子PU/HA复合微球,在静电力作用下进行自组装,并对组装体材料的理化性能进行探究。研究结果表明,当扩链剂物质的量分数为15%,HA质量分数为10%时微球成球性好,粒度分布均匀,具有良好的分散性,组装材料的弹性模量增强,且具有良好的粘弹性。剪切降粘行为表明,在高剪切速率下样品粘度降低,即样品具有一定的可注射性。该自组装凝胶体系具有优良的细胞相容性,为凝胶材料的研究奠定了一定的理论基础,未来有望在骨缺损微创治疗领域得到应用。
[1]YIXue-Min(易学明),LIU Xin-Feng(刘新峰).China Hospital (中国医院),2004,9:7-10
[2]WANG Yong-Guang(王永光).Medicine and Philosophy(医学与哲学),2004,25(11):2-4
[3]Dreifke M B,Ebraheim N A,Jayasuriya A C.J.Biomed. Mater.Res.Part A,2013,101(8):2436-2447
[4]Dou J S,Sprague C H.Chen L,et al.Mater.Technol.,2015, 30(B4):B273-B282
[5]Attenello N H,Maas C S.Facial Plast.Surg.,2015,31(1):29-34
[6]Tommasi G,Perni S,Prokopovich P.Tissue Eng.Part A, 2016,22(11/12):862-872
[7]Werkmeister JA,AdhikariR,White JF,etal.Acta Biomater., 2010,9(6):3471-3481
[8]Hu J G,Guo F,Du J Y,et al.J.Mater.Sci.Mater.Med., 2012,3(23):711-722
[9]JIN Shu-Ping(金淑萍),LIU Ming-Zhu(柳明珠),CHEN Shi-Lan(陈世兰),et al.Acta Phys.-Chim.Sin.(物理化学学报). 2007,23(3):438-446
[10]Wang H N,Hansen M B,Löwik D W P M,etal.Adv.Mater., 2011,23(12):H119-H124
[11]Cheng D,Hou J,Hao L,et al.J.Biomed.Mater.Res.Part B,2016,104(6):1056-1063
[12]Bishop K J M,Wilmer C E,Soh S,et al.Small,2009,5: 1600-1630
[13]Yu L,Zhang Z,Zhang H,et al.Biomacromolecules,2009, 10:1547-1553
[14]Zhang H,Yu L,Ding J D.Macromolecules,2008,41(17): 6493-6499
[15]Mcgarvey J R,Kondo N,Witschey WR T,etal.Ann.Thorac. Surg.,2015,99(2):597-603
[16]Zhao J,Guo B L,Ma P X.RSC Adv.,2014,4(34):17736-17742
[17]XIAO Chao(肖超).Thesis for the Master of Nanjing University of Science&Technology(南京理工大学硕士论文). 2015.
[18]WU Yong-Tao(武永涛).Thesis for the Doctorate of Donghua Unversity(东华大学博士论文).2011.
[19]JIANG Cai-Yun(蒋彩云),WENG Xiao-Lei(翁晓磊),QIAN Wei-Pin(钱卫平).Acta Phys.-Chim.Sin.(物理化学学报), 2008,24(12):2159-2164
[20]LI Chong-Yang(李重阳).Thesis for the Master of Tianjin University(天津大学硕士论文).2006.
[21]FENG Ya-Kai(冯亚凯),WU Zhen-Zhen(吴珍珍).Materials Review(材料导报),2006,6:115-118
[22]Santerre J P,Woodhouse K,Laroche G,et al.Biomaterials, 2005,26(35):7457-7470
[23]Amitai G,Aneersen J,Wargo S,et al.Biomaterials,2009, 30:6522-6529
[24]Howard G T.Int.Biodeterior.Biodegrad.,2003,49(4):245-252
[25]YANG Jian-Jun(杨建军),et al.Polyurethane Medical Materials(聚氨酯医用材料).Beijing:Chemical Industry Press, 2008.
[26]Shanxi Institute of Chemical Industry(山西省化工研究所). Handbook of Polyurethane Elastomer(聚氨酯弹性体手册). Beijing:Chemical Industry Press,2001.
[27]YU Fang-Li(于方丽),ZHOU Yong-Qiang(周永强),ZHANG Wei-Ke(张卫珂),et al.Ceramics(陶瓷),2006,2:7-12
[28]Kikuchi M,Itoh S,Ichinose S,et al.Biomaterials,2001,22 (13):1705-1711
[29]LIAO Jian-Guo(廖建国),LI Yan-Qun(李艳群),DUAN Xing-Ze(段星泽),et al.Prog.Chem.(化学进展),2015(Z1):220-228
[30]ZHU Kai-Ping(朱开平),SUN Jing(孙静),YE Song(叶松), et al.J.Inorg.Mater.(无机材料学报),2016,31(4):434-442
[31]LIU Hao-Huai(刘浩怀),ZHANG Jian-Hua(张建华),XU Qing-Ling(徐庆陵),et al.J.Inorg.Mater.(无机材料学报), 2011,26(10):1073-1077
[32]LING Jian(凌剑),WEI Xiao-Li(卫晓丽),QIU Sheng-Jun(邱圣军),etal.J.North Univ.China:Nat.Sci.(中北大学学报:自然科学版),2006,27(1):59-61
[33]LüQ,Nair L,Laurencin C T.J.Biomed.Mater.Res.Part A,2009,91A(3):679-691
[34]Wang Q,Wang L M,Detamore M S,et al.Adv.Mater., 2008,20(2):236-239
Preparation and Self-Assembly of Oppositely Charged Polyurethane/Hydroxypatite Microspheres
LAI Xin HOU Yi LI Yu-Bao HUANG Min REN Xin ZHANG Li*
(Analytical&Testing Center,Research Center for Nano-biomaterials,Sichuan University,Chengdu 610064,China)
Oppositely charged polyurethane/hydroxyapatite (PU/HA)microspheres were successfully prepared by using the self-emulsifying method,in which 2,2-Bis(hydroxymethyl)propionic acid(DMPA)and N-Methyldiethanolamine (MDEA)was used as chain extender,respectively.The self-assembled gel system was formed by electrostatic force between oppositely charged microspheres.The physicochemical properties of these microspheres were characterized and analyzed by means of SEM,Laser particle size analyzer,ζpotentiometer, XRD and TG,and the rheological properties of the self-assembled system was also studied by rheological test. The results confirm that oppositely charged microspheres are successfully prepared and the best morphology is obtained when the chain extender is 15%(n/n)and HA is 10%(w/w);XRD and TG results show thatthe inorganic phase of HA is successfully incorporated into PU microspheres.The rheological test demonstrate that the selfassembly gel system have higher elastic modulus and injectability.In addtion,the bone marrow mesenchymal stem cells(BMSCs)can adhere,spread and grow well on the surface ofthe material.
anionic PU/HA microspheres;cationic PU/HA microspheres;self-emulsifying;self-assembly;injectable gel
TB33
A
1001-4861(2017)080-1403-08
10.11862/CJIC.2017.164
2017-02-28。收修改稿日期:2017-06-02。
国家自然科学基金(No.51673131,31370971)资助项目。
*通信联系人。E-mail:zhangli9111@126.com
