环境水样中129I分析及其在环境示踪中的应用
2017-07-18姜旭宏侯小琳张路远
姜旭宏,侯小琳,陈 宁,张路远
环境水样中129I分析及其在环境示踪中的应用
姜旭宏1,2,3,侯小琳1,3,陈 宁1,3,张路远1,3
1.中国科学院地球环境研究所 黄土与第四纪地质国家重点实验室,西安 710061
2.中国科学院大学,北京 100049
3. 陕西省加速器质谱技术及应用重点实验室,西安加速器质谱中心,西安 710061
129I是碘的唯一长寿命放射性同位素(半衰期为1570万年),目前环境中129I主要来源于人类核活动,尤其是欧洲核燃料后处理厂的排放。人工129I的大量释放使环境中129I的水平大幅度升高,具有明显的空间分布规律,并且随排放时间呈现明显的变化趋势。129I是一种理想的环境示踪剂,可应用于核环境安全监测,包括核事故监测和评价、核燃料后处理厂129I排放和环境影响评价以及应用于环境过程示踪研究。陆地环境水样中稳定碘(127I)水平较低,129I含量更是处于超低水平,因此,准确分析其中超低含量129I及其化学形态是研究难点。本文系统地评述了环境水样中129I及其形态的分析方法,129I的水平、分布及其在核设施环境安全监测和稳定碘的地球化学循环示踪领域应用的研究进展。
129I;水样;有机碘;形态;环境示踪
1 环境水样中的碘及其同位素
1.1 环境水样中的碘
碘(Iodine,I)是自然界中广泛存在于水圈、岩石圈、生物圈和大气圈中的微量元素。碘的化学性质活泼,易溶于水,在某些植物和动物组织如海藻、甲状腺组织中高度富集,因此碘在环境中分布很不均匀(Hou et al,2009b)。地球上的碘大部分存在于海洋中(Wong and Cheng,1998),约占地球总储量的70%。海水中碘浓度为45 — 60 μg · L−1,陆地环境水样主要包括河水、湖水、大气降水和地下水等,其碘浓度一般远低于海水,其中大气降水中碘含量一般为1 — 6 μg · L−1,地表水(河水、湖水)中碘浓度为1 — 10 μg · L−1(Hou et al,2009b)。
除了不同类型环境水体间的碘含量具有差别,同一类型水体中的碘含量也随地理位置和海拔不同而呈现一定规律。由于碘主要存在于海洋,一般认为陆地上近海地区的碘水平较高。已有研究发现,沿海地区降水中的碘浓度稍高于内地,城区降水中的碘浓度明显高于农村,表明降水碘浓度不仅与海洋补给有关,也与人类活动有关(朱发庆和谭见安,1988)。另外,大气降水中的碘浓度除了随地域变化外,也会随海拔变化出现明显差异。据报道,德国雪水中碘浓度随海拔升高而显著降低,海拔326 m处雪水中碘的平均浓度为4.83 μg · L−1,而海拔1164 m处为1.65 μg· L−1(Gilfedder et al,2007a)。
1.2 环境水样中129I的来源、水平和分布
碘元素共有37种同位素(108I —141I),大多数为短寿命放射性同位素(<1 d),半衰期为10 min以上的碘同位素见表1。127I是碘元素的唯一稳定同位素,而129I是唯一天然生成的长寿命放射性碘同位素,半衰期达1.57×107a。由于其极长的半衰期以及碘元素高水溶性和活泼的化学性质,129I可作为一种良好的环境示踪剂(Edwards,1962)。
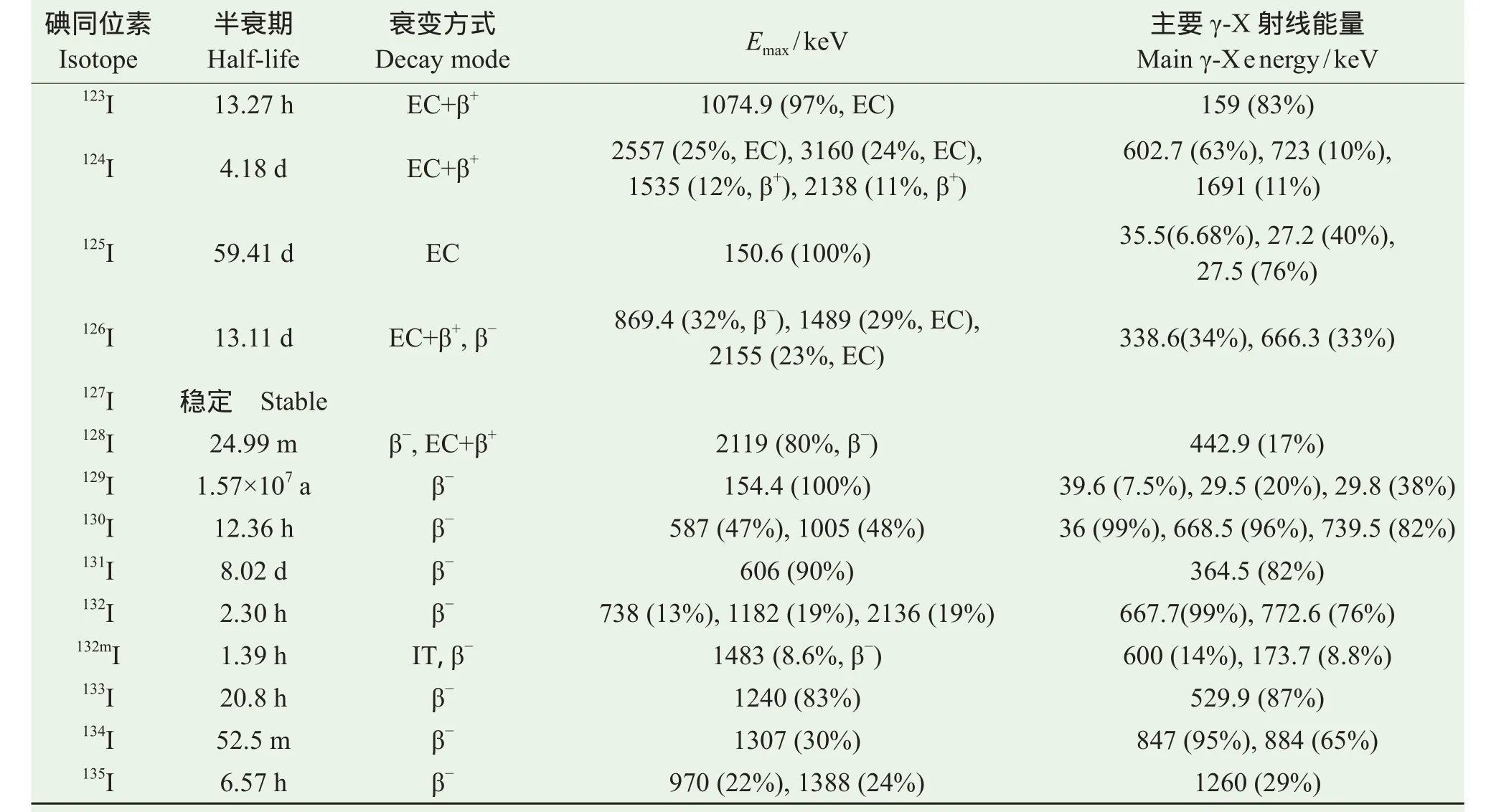
表1 碘的主要同位素及其衰变性质(Hou et al,2009b)Tab.1 Major iodine isotopes and their deday properties (Hou et al, 2009b)
1.2.1 环境129I的来源
虽然129I是一种天然的碘同位素,但目前环境中129I主要来源于人类核活动。天然129I主要由宇宙射线与大气层中的Xe发生散裂反应、地壳中238U自发裂变和235U热中子诱发裂变及Te的中子俘获反应生成(Fehn et al,2007)。其中,宇宙成因和238U自发裂变生成的129I被认为是地球表层库中天然129I的主要来源,并且二者的贡献基本上相当。自然界中天然129I总储量约为5×104kg,其中约99%储存于地壳和海洋沉积物当中,仅有250 kg存在于地球表面的水圈、岩石圈和大气圈中(Fabryka-Martin,1985),而其中约有140 kg存在于水圈中(Snyder et al,2010)。环境中天然源和人为源的129I的产生量见表2。
碘在海洋中的滞留时间约为40000 a,远大于全球海洋水体的交换时间(~1000 a),因此通常认为海洋系统中天然碘同位素是均匀分布的(Fabryka-Martin,1985;Moran et al,1998)。相比碘在海洋-大气间的交换速率,大气中宇宙成因129I的生成速率非常缓慢,因此一般认为大气和海洋中的碘同位素处于平衡状态,整个水圈在核前时期的碘同位素比值应该是一致的(Moran et al,1998)。基于这个前提,前人通过分析海洋沉积物获得的核前海洋环境中129I/127I原子比值为1.5×10−12,该值被用做海生环境129I/127I的初始值,广泛用于海生环境样品的年龄测定(Edwards,1962;Fabryka-Martin,1985;Fehn et al,1986,2000;Moran et al,1998)。陆生环境129I/127I的初始值研究较少,目前已有报道黄土高原深层(深度为63.5 m)黄土中天然129I/127I比值为20×10−12(Hou et al,2010),比海洋环境中的129I /127I初始值高一个数量级,认为这可能是由于陆生环境稳定碘(127I)含量较低(如土壤中碘的浓度在0.5 —40 μg·g−1,大部分在1 — 3 μg · g−1)所致(范煜坤,2013)。同时,研究发现红黏土中天然129I/127I比值为5.34×10−12,约为黄土的四分之一,这一比值与海洋环境初始值较接近(范煜坤,2013)。
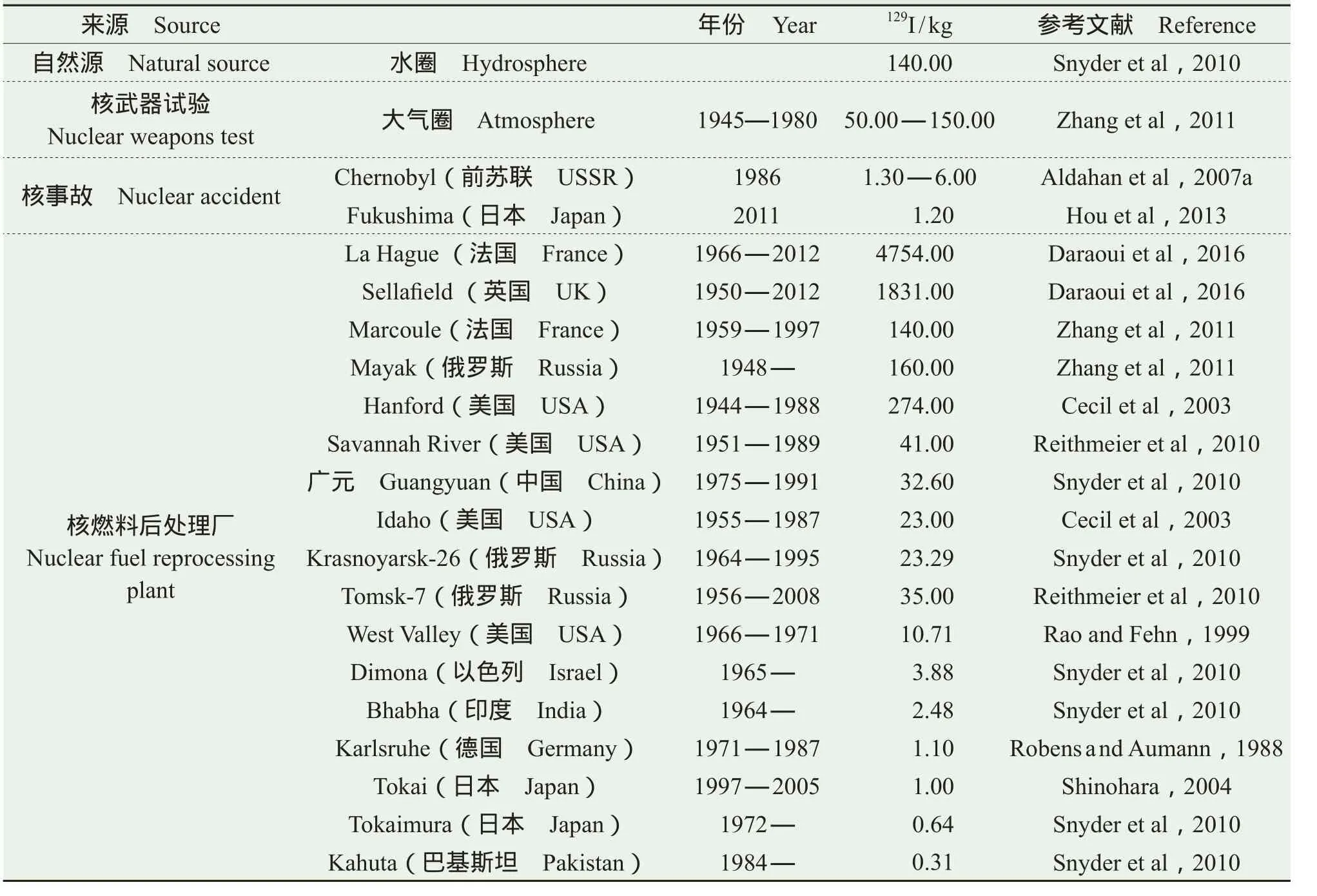
表2 环境中129I主要来源及其排放量Tab.2 The main source of129I in the environment and its emissions
20世纪40年代以来,人类核活动向环境释放了大量的129I,使环境中的129I水平远高于天然水平(Raisbeck et al,1995)。人类核活动产生的129I主要通过235U和239Pu经过裂变反应产生,由核燃料后处理厂、核武器试验、核事故以及核反应堆运行中的泄漏排放等4种途径释放于环境。(1)核燃料后处理厂排放是最重要的释放途径,法国的La Hague和英国的Sellafi eld核燃料后处理厂是目前世界上两个最大的核燃料后处理厂,截止到2012年两者的129I总排放量已超过6500 kg,占全球核活动129I总排放量的90%以上(Raisbeck et al,1995;Daraoui et al,2016)。(2)据估计,1945—1980年全球大气核武器试验累计向环境中释放了50 — 150 kg的129I (Zhang et al,2011),这些129I除进入大气对流层的部分在局部区域沉降外,大部分进入平流层,长期滞留并混合均匀后扩散进入对流层,沉降到地表,造成全球范围内129I水平的升高。研究表明大气核武器试验使北半球中纬度地区129I/127I水平比核前水平提高了1—3个数量级,使南半球129I/127I水平比核前水平提高了1—2个数量级(Rao and Fehn,1999;Reithmeier et al,2006;Snyder et al,2010; Xing et al,2015)。(3)迄今为止全球共发生了4次较大的核反应堆事故(1986年4月26日切尔诺贝利核电站反应堆事故,2011年3月11日日本福岛第一核电站事故,1957年10月10日英国温茨凯尔核事故和1979年3月28日美国三里岛核事故),其中切尔诺贝利事故和福岛核事故分别向环境释放了1.3 — 6 kg和1.2 kg的129I(Aldahan et al,2007b;Hou et al,2013)。已测得切尔诺贝利核事故高污染地区环境样品中129I/127I比值高达10−6(Duffa and Fréchou,2003;Reithmeier et al,2006);日本福岛核事故使福岛当月收集到雨水中的129I/127I比值从核事故前的10−8增加到10−5(Xu et al,2013);(4)核反应堆运行过程中虽然会产生并可能排放少量的129I,但目前尚未有核反应堆运行造成环境129I水平升高的报道。He et al(2011)和张鸿等(2012)测定了中国核电站附近表层海水中129I的水平,其129I/127I比值处于10−11—10−9,与同纬度其它本底地区没有明显差别,表明核电站对周边海洋环境中129I影响甚微。
1.2.2129I的环境水平及其分布
由于上述人为排放源的位置及排放量不同,环境中的129I水平和分布极不平衡,目前已报道的环境样品中的129I/127I比值差异较大。大气核武器试验沉降导致全球表层环境中129I/127I比值从10−12升高至10−11—10−9,而在核燃料后处理厂周围以及核事故污染地区,129I/127I比值可高达10−6—10−4。根据人类核活动的污染程度不同,环境中的129I水平可分为:未受污染的核前水平(10−11—10−12);核活动轻微污染的水平(即全球沉降的环境背景水平,10−11—10−9);受核活动直接污染区域的水平(10−9—10−6)以及核设施附近或核活动高污染区域的水平(10−6—10−3)(Hou et al,2009b)。环境本底区域水样(湖水、河水、降水)中129I/127I比值一般为10−9—10−7,而收集于核燃料后处理厂和核事故周围地区的水样中的129I水平显著升高。同一地区不同类型的水体也有一定差异,一般降水中的129I/127I比值高于地表水(河水和湖水) 的129I/127I比值 (Santschi and Schwehr,2004)。
图1总结了目前已报道的降水中的129I数据,采样点主要集中在北半球,南半球可获得的数据极少,只有南极雪水的少量数据,且所选择数据主要为非特殊时期(如核事故等)的日常环境监测值。降水中的129I高值点主要出现在有点源排放的地区,包括法国La Hague、英国Sellafi eld、和日本Tokai核燃料后处理厂周围,且欧洲降水中129I水平明显高于日本,这是由于La Hague和Sellafi eld的129I排放量相对巨大的缘故。在欧洲,中欧(如德国、爱尔兰)降水中的129I水平最高,浓度高于1010atoms · L−1,129I/127I比值高达10−6;其次为北欧(瑞典、挪威、芬兰、丹麦),129I浓度为108—109atoms · L−1,129I/127I比值为10−7—10−8;南欧(如西班牙、意大利)最低,129I浓度为107—108atoms · L−1,129I/127I比值为10−8—10−9。日本降水中的129I水平比除欧洲以外世界其它地区高1—2个数量级,主要为Tokai核后处理厂少量的气态排放及福岛核事故的一些影响。美国虽然有核后处理厂,但由于采样点与排放源距离较远,降水中的129I浓度水平为107atoms · L−1,129I/127I比值为10−9,与北半球其它地区一致,反映的是核武器试验和核燃料后处理厂排放的129I全球沉降水平。南极地区降雪中的129I浓度为106—107atoms · L−1,129I/127I比值为10−9—10−10,比海洋初始值高了2 — 3个数量级,主要是大气核武器试验释放129I的长距离传输引起的(Xing et al,2015,2016)。而我国降水中129I水平研究很少,2011年报道了西安地区129I浓度为1.03× 108atoms · L−1,129I /127I比值为2.12×10−9(Zhang et al,2011),与北美水平较为一致。本实验室正在进行西安地区长时间序列(2008年以来)降水中的129I分析,得到的129I浓度平均值处在10−8—10−9范围内,基本处在129I全球沉降水平(未发表)。
Snyder et al(2010)总结了全球表层水(600多条河流,湖泊和浅层海水)中的129I浓度分布,得到了较全面的环境水体129I分布情况,尤其是包括了赤道地区和南半球的信息。北半球地表水中129I浓度分布显示:在距离核后处理厂较近区域水平明显较高,用差值方法得到的分布图显示赤道地区和南半球地表水中129I水平较低,为106—107atoms · L−1,且南半球水平低于赤道附近地区。Snyder and Fehn(2004)对收集于全球不同地区的地表水中129I分析也表明,南半球的129I水平比赤道地区稍高,且129I/127I比值在南半球中纬度地区出现一个高值点。这些都表明从排放源释放的129I可以在全球范围内实现快速的大气传输。最新研究表明,南半球阿根廷河水和湖水中129I浓度为(1.44—7.5)×105atoms · L−1,处于天然水平(3.7×104atoms · L−1) 和大气核武器试验沉降水平(1.0×106atoms · L−1)之间;其129I/127I比值为(0.57—6.7)×10−11,比南极雪水的129I/127I比值(6.8 — 9.5)×10−10低一个数量级(Negri et al,2013;Xing et al,2015)。加拿大西北靠近北极地区的河水和溪水中129I浓度为106— 107atoms · L−1,129I/127I比值为10−9—10−10,比北半球其他地区至少低一个数量级(Herod et al,2013)。
总的来说,陆地环境水体中129I水平的分布特征为,北半球中高纬度范围靠近排放源的地区最高,其它纬度范围远离排放源地区因受到排放源不同程度的影响相对较高;赤道、南半球和南极地区处于较低水平,主要129I来源为大气核武器试验的全球沉降。
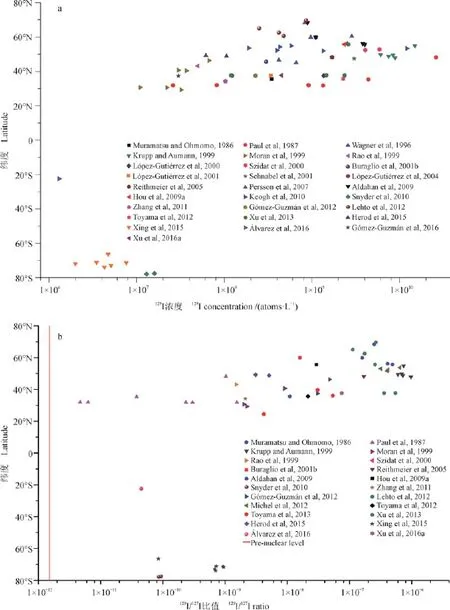
图1 全球不同地区降水中129I浓度(a)和129I/127I原子比值(b)随纬度的变化Fig.1129I concentration (a) and129I/127I atomic ratio (b) in precipitation as functions of latitude
由于20世纪开始的各类核活动排放的129I是不断变化的,因此环境中129I水平除了具有空间分布的差异外,也具有时间上的差异性。大气核武器试验129I排放主要发生在20世纪50 — 60年代,核后处理厂从20世纪50年代开始持续向环境中释放129I,并且在20世纪90年代排放量迅速增加,这些都导致环境中129I水平随时间的变化。图2为欧洲阿尔卑斯山Fiescherhorn冰芯中20世纪50年代以来的129I沉降量随时间的变化和日本四个不同地点(Tokyo、Tsukuba、Akita和Ishigaki)1963—2005年降水中129I沉降量的变化,可以看出,从20世纪50 — 60年代开始,欧洲冰芯中129I沉降量开始缓慢增加,这一阶段主要受全球核武器试验的影响,之后出现短暂降低后再不断升高,于1980 — 1990年达到最高,这段时间主要是受到欧洲核燃料后处理厂大气排放的影响。阿尔卑斯山距离欧洲核后处理厂较近,其129I沉降量主要受到法国La Hague、英国Sellafield及法国Marcoule(1959—1997年)的影响(Wagner at al,1996;Reithmeier et al,2006)。日本几个采样点中Tokyo和Tsukuba由于距离日本Tokai核后处理厂较近,其129I沉降量与该处理厂的气态排放量直接相关;Akita位于Tokai核后处理厂主风向的上风向,受该处理厂排放影响较小,其129I沉降量的变化与阿尔卑斯山冰芯中的变化相似,表明其可能与欧洲核后处理厂的气态排放有关;而Ishigaki采样点位于太平洋,129I沉降量比其他地区都要低,推断其129I更多的来源于海洋气团,而不直接受到日本或欧洲核后处理厂的影响(Toyama et al,2012,2013)。
1.3 环境水样中碘同位素的化学形态
碘具有−1,0,+1,+3,+5和+7价等多种化学价态,在不同条件下可形成碘的不同存在形态。水体中的无机碘主要为I−和,其它价态如I2,OI−和在环境水样中不稳定,难以存在。由于碘具有亲生物性,除无机碘外,环境中还有大量有机碘,包括挥发性的烷基碘、水溶性的小分子有机碘以及蛋白质、酚类化合物、腐殖质等大分子中的有机结合态碘等(Hou et al,2009b)。环境水样中碘的形态与其氧化还原环境和生物的活动密切相关。在海水中,碘主要以、I−和小分子有机碘形式存在,而在海岸区域,由于生物的作用,海水中的有机碘含量升高,可占总溶解态碘的5% — 40% (Wong,1991)。海岸区域有机碘组成主要为小分子气态碘化物,如CH3I、CH2ClI、CH2I2、CH3CH2CH2I,尽管海水中有机碘浓度较低,却在碘的循环中具有重要作用。碘从海洋传输到陆地环境,主要是通过有机碘化合物从海水中释放实现的(Hou et al,2009b)。陆地环境水体中的碘也主要以I−,和有机碘形式存在,但有机结合态碘的含量比海水中的较高,且不同类型水体中碘的形态差异很大。研究发现河水中有机碘含量占比为35% — 75%(Oktay et al,2001;Schwehr and Santschi,2003),湖水中有机碘含量占比为85%(Gilfedder et al,2009),而降水中有机碘含量占比甚至高达94%(Gilfedder et al,2007b,2008)。
关于环境水样中129I的形态研究较少。据报道德国1994—1995年雨水中129I主要形态为无机碘,约占总129I的43.8% — 79.1%,有机碘占比为20.9% — 56.2%(Krupp and Aumann,1999);丹麦降水中129I的主要形态为I−,127I的主要形态为(约占总127I的65%),非离子态碘的占比不到10% (Hou et al,2009a);Lehto et al(2012)测定了芬兰湖水和雨水中的129I和127I形态,发现这两种同位素的主要形态均为I−,且129I中I−形态的比例比127I的高,湖水中129I−的比例比雨水高。
环境中痕量元素的性质与其赋存的状态密切相关,元素存在的形态不同,其性质会相差很大(何红蓼等,2005;刘崴等,2008)。环境中的碘及129I以不同的形态存在,它们的化学行为也不相同。环境中的129I的形态取决于它的来源,并随环境因素发生变化,因此可用来进行环境过程研究(Fan et al,2013)。例如通过分析丹麦地区降水中129I和127I不同的化学形态,可以获得其129I和127I不同的来源,并可用于探究碘的大气化学过程及地球化学行为 (Hou et al,2009a)。
环境水样中碘的含量较低,而129I/127I比值一般为10−6—10−9,129I含量与127I含量相差数个数量级。此外,碘由于形态多样,且淡水中有机碘含量高,因此准确分析环境水样的129I含量及各形态分布更是一项巨大的挑战。随着分析技术与方法的发展,目前已形成了一系列环境水样中129I及形态的分析方法。
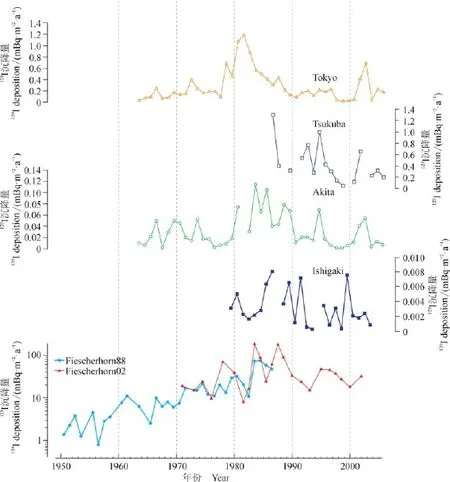
图2 欧洲Fiescherhorn冰芯和日本Tokyo、Tsukuba、Akita和Ishigaki地区大气沉降129I通量随时间变化(数据来源于Wagner et al(1996),(Reithmeier et al,2006),(Toyama et al,2012,2013) )Fig.2 Temporal variation of129I deposition fl ux in Fiescherhorn ice core and in precipitation from Tokyo, Tsukuba, Akita and Ishigaki, Japan (Data from Wagner et al (1996), Reithmeier et al (2006), Toyama et al (2012, 2013))
2 环境水样129I的分析方法
对环境水样中的129I进行分析,首先需要确定适合的129I测量方法,然后根据测量技术的要求,分离样品中的碘,并制成适合的靶源。由于环境样品中129I的浓度极低,因此一般需要进行分离和浓集操作,富集水样中的碘并纯化去除干扰后测量,以获得需要的探测限和分析准确度。环境水样尤其是陆地水样中,有机碘占总碘份额很大,在进行总129I分析时,需先将有机碘转化为无机碘,然后采用无机碘分析方法进行分离、制备和测量。常见的129I分析流程包括样品预处理、有机碘的分解、分离富集纯化、靶源制备和129I测量。对于环境样品中稳定碘和129I形态分析方法,也进行了总结。以下分别总结本实验室及其他相关研究在129I化学分析方面取得的进展。
2.1129I的测量方法
129I 是一种β衰变的放射性核素,释放出能量为150 keV的β射线,同时发射39.58 keV的γ射线以及29 — 30 keV的X射线(Hou et al,2009b)。因此,γ和X射线能谱及液体闪烁计数(LSC)等放射性测量技术可对其进行测量(Sutiez et al,1996;Bouisset et al,1999)。但由于129I具有极长的半衰期,这两种技术的灵敏度和探测限均较差,难以用于环境水平129I的分析(Hou et al,2009b)。中子活化技术(NAA)也可用于129I测定,其原理是基于中子活化129I使其生成短寿命130I,使用γ能谱仪测量其高能γ射线(536 keV(99%),668.5 keV(96%),739.5 keV(82%))实现129I的定量,从而提高129I的分析灵敏度。但中子活化时样品中的其它元素也会发生活化反应,产生的放射性核素会严重干扰130I的测量,因此通常需要对样品中的碘进行预浓集和分离,并在中子照射后再次对样品中的碘进行放射化学分离纯化,流程十分繁琐耗时,而且该技术最大的局限性在于样品中大量的127I可能通过连续中子活化生成130I,干扰129I的测定,因此无法用于129I/127I比值低于10−10的样品中129I的测定(Hou et al, 2009b)。质谱技术,包括加速器质谱(AMS) 和电感耦合等离子体质谱(ICP-MS),是20世纪80年代后发展并开始用于129I测量的技术,其通过对原子直接计数进行测量,因此特别适合于长寿命核素的测量(Cox and Pickford,1992;Brown et al,2007)。在ICP-MS测量中,由于稳定同位素129Xe的强烈干扰和较低的碘的离子化效率,该技术对129I的探测极限难以满足一般环境样品的分析需求(Hou et al,2009b)。AMS是目前最为灵敏的129I测量技术(Buraglio et al,2000),且129Xe在离子源中不会生成负离子而对129I的测量产生干扰,其它干扰可以通过加速管剥离去除分子离子和之后的碘离子的价态选择消除,因此该技术可以探测129I/127I比值低至1×10−14水平的129I (Hou et al,2009b)。表3比较了上述几种129I的测量技术的主要特性。
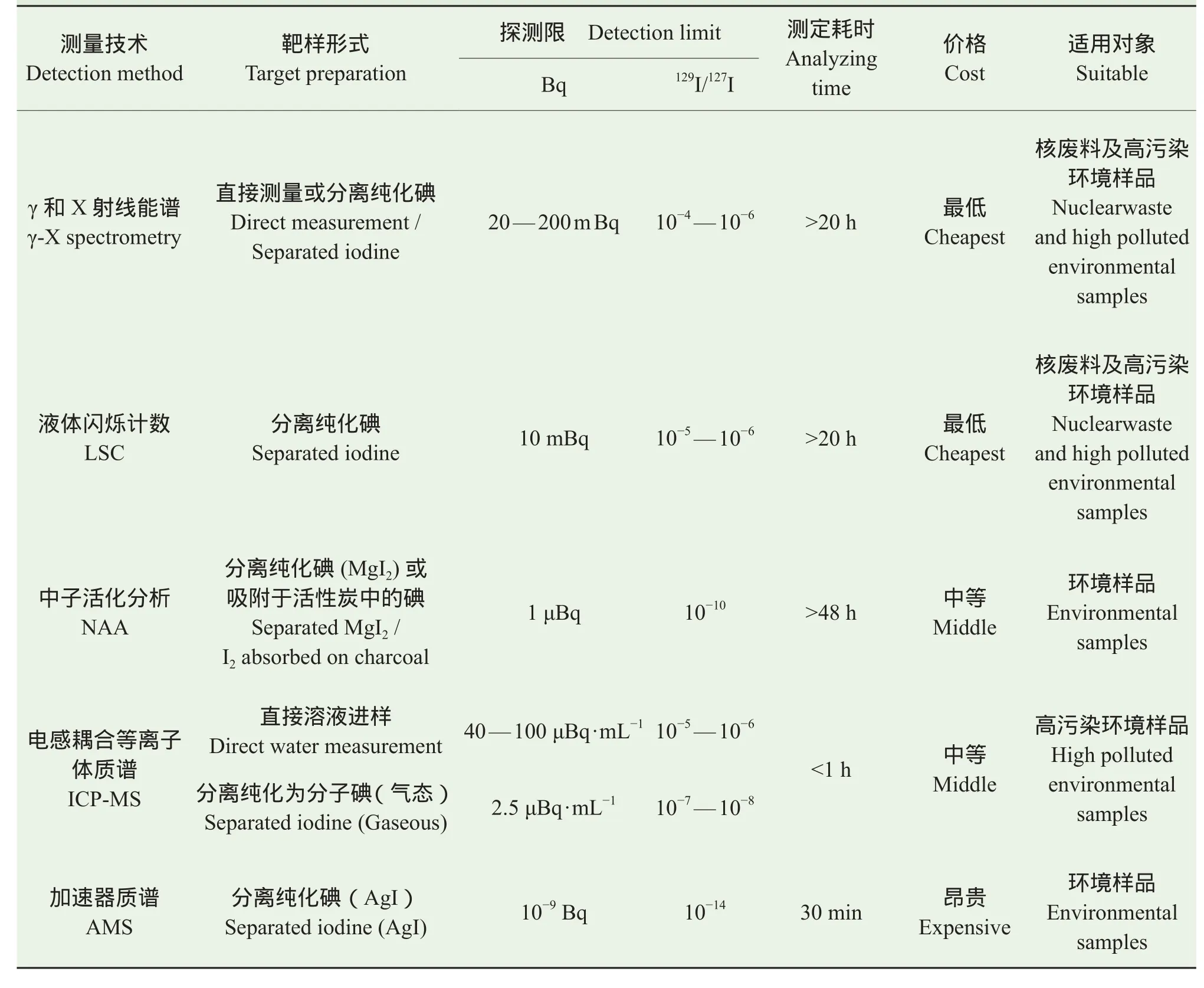
表3 129I测量技术及其特点(Hou et al,2009b)Tab.3 Comparison of129I determination methods (Hou et al, 2009b)
2.2 环境水样中有机碘的分解方法
陆地环境样品中的碘除了以I−和形式存在外,还有大量的有机碘(Schwehr and Santschi,2003)。过去的分析方法不能提取有机碘,使129I被低估,所以发展有机碘的分解方法是一直受到关注的问题。目前129I分析方法常在分离富集步骤之前加入有机碘的分解步骤,即先选用适合的方法将有机碘分解,使之转化为无机碘,再进行后续分离、测定步骤。
目前已报道的有机碘分解方法包括以下几种,(1)紫外(UV)灯照射:将样品在1200 W的紫外(UV)灯下照射3.5 h以上,将有机碘转化为无机碘(Reifenhauser and Heumann,1990);(2)高温热解法:将小体积水样蒸干后与催化剂混合后置于管式炉中经一定升温程序至900℃,将有机碘转化为无机碘并收集于碱性溶液或活性炭中(Zhang et al,2010);(3)碱消解法:样品溶液加入NaOH(NaOH浓度0.30 mol · L−1),然后在120℃的加热板上加热3 h将有机碘分解为无机碘(López-Gutiérrez et al,2001;Hou et al,2009a;Xu et al,2013);(4)超声碱消解法:样品中加入NaOH和乙醇,转移到聚四氟乙烯罐中,在65℃下超声消解3 h后继续反应过夜,然后再次加入NaOH和乙醇并重复超声,再放在加热板上65℃下加热0.5 h,使有机碘转化为(Schwehr and Santschi,2003);(5)氧化消解法:在pH 1—2和60℃条件下,使用强氧化剂K2S2O8分解水样中的有机质,并把有机碘转化为形式(Dang et al,2013;Xing et al,2015)。
表4比较了上述几种方法对于水样中有机碘分解的优缺点及适应范围。这几种方法中仅碱消解法和氧化消解法能够用于处理大体积环境水样(>1 L),但碱消解法对于水样中的有机大分子如腐殖质的分解能力不足,可能难以完全分解水样中的有机碘。研究表明氧化消解法可完全将各种水样中的有机碘转化为无机碘(Dang et al,2013)。
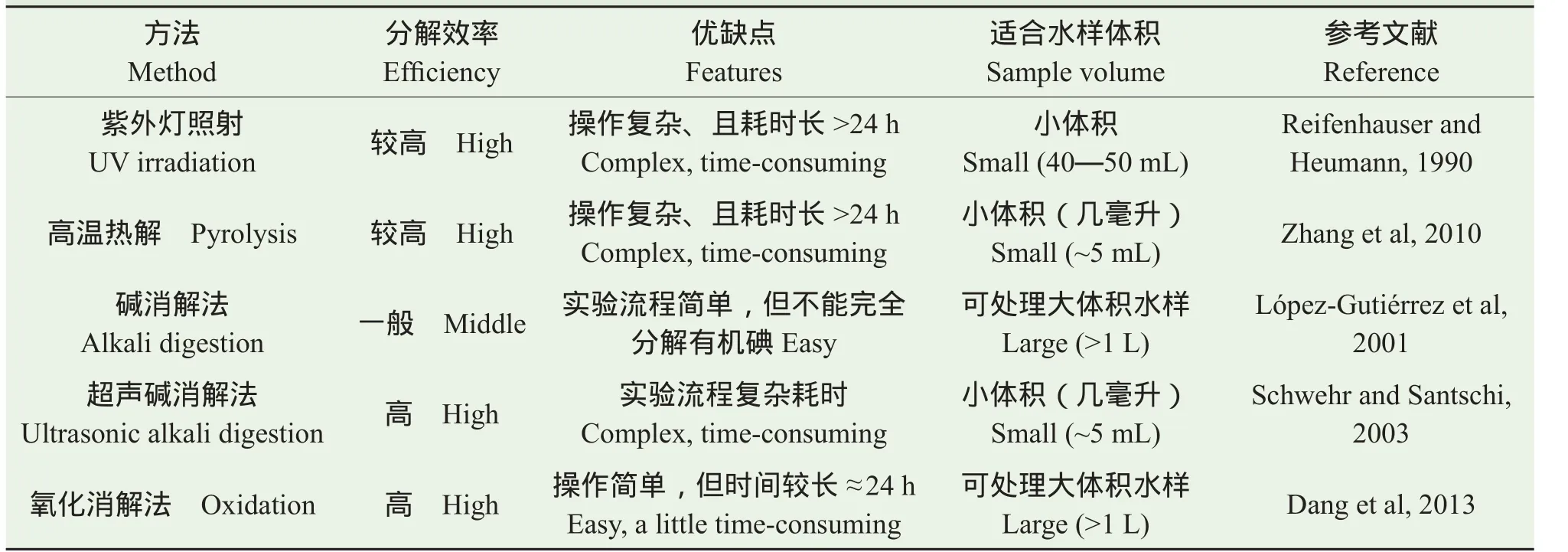
表4 水样中有机碘分解方法及其特点Tab.4 Comparison of organic iodine decomposition methods
2.3 水样中总无机碘的富集分离方法
环境水样中的无机碘以及转化为无机碘的有机碘的分离富集主要有以下四种方法:
(1)AgI直接沉淀
对于129I含量较高的小体积水样,先加酸调节至pH<2,再加入少量NaHSO4还原为I−(如果其碘含量低于0.05 μg · L−1,可先加入适量(0.5 —2 mg)的127I载体),最后加入AgNO3生成AgI沉淀后进行测定。该方法具有回收率高、耗时短、操作简单等优点,适用于小体积水样,如放射性废水和受核活动直接影响的水样的分析( 罗茂益,2013)。但样品中含有的、Cl−、Br−等离子易与Ag+反应生成大量的沉淀,影响后续129I的测量,需使用HNO3和NH3· H2O洗涤去除这些杂质。
(2)溶剂萃取
根据非极性I2与有机溶剂CCl4或CHCl3相似相溶原理建立的溶剂萃取方法是目前最为常用的分离、富集、纯化水样中碘的方法。在有机碘转化后的50 — 1000 mL水样中加入1 — 2 mg的载体碘和适量125I示踪剂,用HNO3调节至pH<2,再加入适量的NaHSO3将还原至I−。然后加入30 — 50 mL CCl4,再加入适量NaNO2或H2O2氧化I−为I2,将其萃取到CCl4相。用NaHSO3还原并反萃至水相。重复上述萃取与反萃取过程直至反萃液体积<5 mL。最后向反萃水相中加入AgNO3生成AgI沉淀后测定。该方法的回收率与样品体积、碘浓度、样品盐度等有关,通常可达到95%以上,且最终制得的靶样中只含AgI,无其他杂质,测定时计数稳定。已被广泛应用于水样总无机碘的分离制备,特别适用于体积<1 L的水样,如表层环境水样品及油田卤水样品中碘的分离制备(Buraglio et al,2001a;Herod et al,2013;Chen et al,2014)。但该方法操作过程中所使用的有机溶剂有毒,且极易挥发,因此必须在通风橱中操作。
(3)AgCl-AgI共沉淀
根据Cl与I相似的化学性质,Hou et al(2010)创新性地建立了AgCl-AgI共沉淀方法分离、纯化、富集碘,并用于加速器质谱测定129I。首先用HNO3调节溶液至pH<2,加入适量的NaHSO3将还原至I−,并加入0.5 mg Cl−和适量的AgNO3,生成的大量AgCl和Ag2SO3载带样品中微克量级的AgI沉淀。然后使用HNO3洗去Ag2SO3和Ag2SO4沉淀,再依次用10%和5%的氨水洗涤沉淀,以去除过量的AgCl,并控制沉淀质量至1—3 mg。该方法已成功用于大体积(<50 L)的水样(海水、淡水),离子交换预浓集后的洗脱液以及固态样品热解后的碱性捕集液和固体样品浸取液中微克量级的碘的分离,并成功用于分离和分析南极雪水样品低水平129I(Hou et al,2010;Xing et al,2015)。该法的主要优点是无需加入稳定碘载体(127I),避免了稳定碘中含有的少量129I对于低水平样品中超微量129I分析的干扰。
(4)离子交换
对于大体积、低129I水平水样中碘的分离富集,可以采用离子交换法实现预浓集,有批式和柱式两种实验方式。首先向水样中加入适量碘载体,并加入适量的NaHSO3将还原至I−,混合均匀。批式法是直接向溶液中加入处理好的AG1×4 Cl型强碱性阴离子交换树脂(50 — 100目),机械搅拌30 min,待树脂下沉后将水样转入另一容器,再加入树脂搅拌20 min;合并两次吸附碘的树脂,将树脂转入解吸器内,加入8.0%的NaClO,搅拌30 min,重复解吸两次,合并解吸液;将解吸液中的碘调整为I−形态,再采用溶剂萃取的方法完成萃取、反萃、沉淀制靶等步骤。该方法碘的回收率为60%以上,主要用于大体积水样(>100 L)中碘的富集,但分离后的碘仍需进一步浓缩,操作较复杂(谢运棉等,1998)。柱式法是将样品溶液以2 mL · min−1的流速加载到阴离子交换色谱柱上,后采用2 moL · L−1NaNO3或5%—10% NaClO溶液洗脱碘离子,该方法的回收率可以达到80%以上,适用于中等体积水样(如水样体积<1 L的海水样品)的富集浓缩,虽然可以处理大体积水样,但耗时较长(Hou et al,1999)。
(5)银粉吸附
根据单质I2可以与银粉直接反应生成AgI的机理,Yiou et al (2004)建立了无载体碘的银粉吸附制备方法。在<100 mL的海水中加入HNO3和3—10 mg的Ag粉,搅拌24—48 h,制成AMS测量所需的AgI靶样。该方法流程简单、易操作,适用于低127I含量和低129I/127I比值样品(比如未受核活动污染的深层海水)的分析,可避免载体中的129I对样品中129I测量的影响(Yiou et al,2004)。然而该方法对碘的回收率较低,仅为30%(Hou et al,2010)。表5比较了上述几种无机碘分离富集纯化方法的主要特点。
2.4 水样中碘同位素的化学形态分析方法
2.4.1 稳定碘(127I)的形态分析方法
电感耦合等离子体质谱(ICP-MS)技术发展迅速,已成为环境水样中稳定碘(127I)测定的主要方法,其与分离技术联用为形态分析提供了强有力的检测手段。目前环境水样中127I不同形态的分离技术主要包括气相色谱法(GC)和高效液相色谱法(HPLC)。GC-MS以气相作为流动相,需要将样品通过高温转化为气相后进样。与GC相比,HPLC在室温下进行,更适合环境样品的分析(何红蓼等,2005)。HPLC中的离子色谱(IC)是应用较多的分离技术,与ICP-MS连用的方法已成功应用于海水、降水、河水和卤水中稳定碘形态的测定(Yoshida et al,2007;Zheng et al,2011)。Schwehr et al(2003)报道了采用粒度为13 μm的250 mm×4 mm Dionex AS11强阴离子交换柱,并在其前加50 mm×4 mm Dionex AG11保护柱的HPLC-ICP-MS联用技术,测定淡水和海水中的I−、及有机碘的方法,检出限可达0.2 μg·L−1。Zhang et al(2010)报道了利用GC-MS方法测定高碘含量地下水中碘的形态,其使用TR-5MS毛细管柱(30 m×0.25 mm,0.25 μm)进行分离,先测定I−浓度,并通过测量Na2S2O5氧化前后溶液中I−浓度的差异来计算浓度,然后在900℃下燃烧将无机和有机碘转化为后测定总碘,有机碘通过总碘和总无机碘(I−和)之差计算得到。碘化物和碘酸盐的检出限分别为0.04 μg·L−1和0.14 μg·L−1(Zhang et al,2010)。
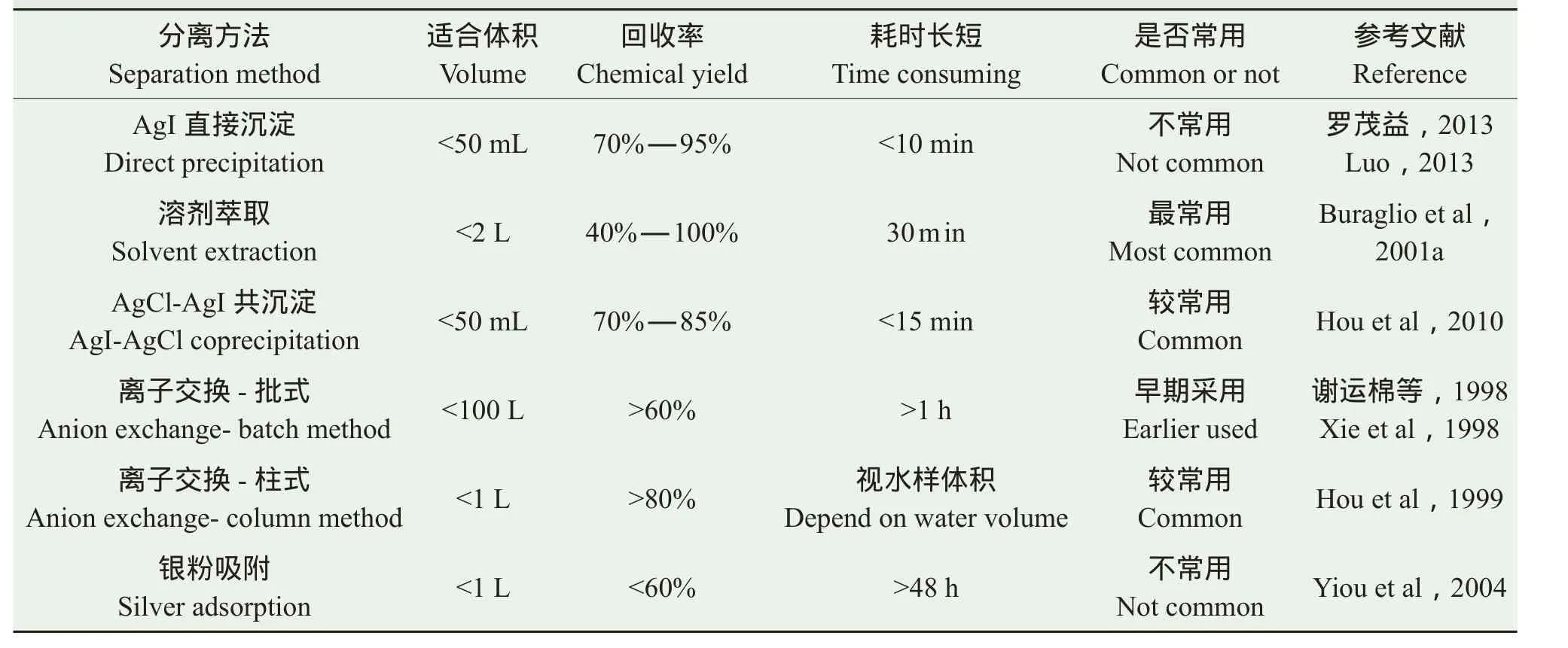
表5 水样中碘的分离和富集方法及其特点Tab.5 Comparison of separation and enrichment of iodine in water samples
2.4.2129I的形态分析方法
虽然可采用GC-MS方法直接测定水样中129I,但该方法仅适用于高污染地区的水样(如萨瓦纳河地区,129I/127I >10−3)(Zhang et al,2010)。对于一般环境水样,其129I水平通常在10−12—10−15g · L−1的范围内,上述稳定碘的形态分析方法难以直接用于129I的形态分析,需要先将不同形态的129I从大体积样品中分离并富集浓缩,然后用高灵敏度的分析方法(AMS和NAA)分别测定各组分中的129I。
离子交换是碘形态分离的常用方法,原理是基于强碱性阴离子交换树脂对I−的亲和力远高于和水中其它阴离子,将I−,和有机碘从水样中分离。为了提高色谱柱对水样特别是海水样品中碘的分离容量,需要用2 mol · L−1NaNO3将树脂由Cl−型转化为型。由于有机碘在离子交换树脂上的吸附行为复杂,难以直接分离 (Hou et al,2001),通常将过滤后的水样直接上离子交换柱,I−吸附在树脂上,而和大部分有机碘流出。用0.5 mol · L−1NaNO3和水淋洗后,用2 mol · L−1NaNO3或5% NaClO洗脱柱上的I−。对于的分离,在流出液中加入NaHSO3溶液并调节pH<2还原为I−,再上离子交换柱,用相同方法淋洗和洗脱,获得的洗脱液为。对于有机碘的分离,可以先将水样中的有机碘转化为无机碘,然后采用溶剂萃取或将所有碘转化为I−后用离子交换色谱的方法分离,获得总129I的量,扣除I−和的量得到有机碘的量(Hou et al,2009a)。分离所得各组分中的碘再进一步采用溶剂萃取分离和富集并转化为I−,最后加入AgNO3制成AgI靶源,用于加速器质谱测定129I,获得129I的化学形态。该方法已成功应用于海水、降水、湖水等样品,特别是低129I含量、大体积的水样中129I的形态分析 (Schwehr et al,2005;Lehto et al,2012)。
除了实验室常规形态分析方法,快速现场分析方法也是一个重要的技术问题。为了就地分离海水中的碘形态,目前在无载体AgI-AgCl共沉淀法基础上,Luo et al(2013)报道了一种简便的选择性共沉淀方法。该方法通过严格控制pH(4.2 — 5.5)和NaHSO3浓度以及加入的AgNO3量,可利用AgI-AgCl共沉淀选择性分离I−,而使留在溶液中,从而实现二者分离。该方法对I−的分离效率大于95%,并且129和129I−组分之间的交叉污染小于3%,是一种快速简单的水样中129I形态分离的方法(Luo et al,2013)。该方法适合含Cl−较高的海水,但不太适合淡水样品和低盐度水样。以上方法只能分离水样中I−和,对于环境水样129I形态中有机碘部分的分离,可以将水样中的有机碘转化为无机碘形态后通过萃取和沉淀分离获得总碘,扣除I−和后的差值为有机碘含量。这一方法可实现在野外采样船上进行快速现场预处理,对于野外工作十分有意义。
图3概括了目前环境水样中碘同位素(129I和127I)及其形态分析的主要方法。
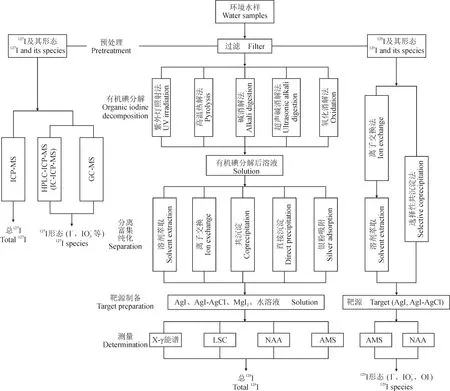
图3 环境水样中碘同位素(129I,127I)及其形态分析方法(改自Zhang and Hou(2013))Fig.3 Schematic diagram of procedures for determination of iodine isotopes (129I,127I) and their chemical species in environmental water samples (Modifi ed from Zhang and Hou (2013))
3 水样中129I及其形态分析的应用研究
核武器试验、核反应堆运行、核事故泄漏和核乏燃料后处理厂等向环境排放了大量的129I,这些129I进入到环境中后会参与环境过程,并会通过食物链进入到人体。由于其极长的半衰期和较低的放射性活度,目前环境129I水平(129I/127I <10−6)对人体造成的辐射危害(<1 μSv/y)远低于天然本底放射性的辐射水平 (>0.1 mSv/y),没有显著的辐射影响(Hou et al,2009b)。但通过分析环境样品,包括水样中的129I及其形态,可以示踪研究各种环境过程。近年来,人工129I的环境示踪得到广泛应用,取得了一系列成果。
3.1 核设施环境安全监测
目前国际上很多国家已开展了129I核安全示踪研究,包括欧洲、北美、亚洲等多个国家,主要集中在核事故影响水平评估、短寿命高危害131I的水平和辐射影响重建、以及核设施的环境安全示踪研究等。
3.1.1 核事故监测和评价
作为235U和239Pu的挥发性裂变产物,在核事故中,129I会与其它挥发性裂变和活化产物一起释放到环境中,包括131I、132I和133I以及其它挥发性放射性核素如137Cs、134Cs。在所有释放的核素中,131I具有较高的裂变产额(2.83%),且碘能够在人体内(甲状腺)高度富集,所以131I的辐射危害最为显著。而131I的半衰期只有8 d,很快会衰变殆尽,难以进行完整的跟踪监测。由于129I和131I具有相同的生成路径和环境化学行为,因此可用129I的分析来重建核事故发生时环境131I水平以及辐射影响等(Paul et al,1987;Straume et al,1996)。
1986年的切尔诺贝利核事故释放了大量的131I以及其它放射性核素。据报道,俄罗斯、乌克兰和白俄罗斯高度污染地区儿童的甲状腺癌发病率显著增加。Paul et al(1987)分析了切尔诺贝利事故后一个月采集于德国慕尼黑和以色列的雨水样品,测得其129I含量分别为2.6×1010atoms · L−1和1.2× 109atoms · L−1。以1982年以色列雨水样品中的129I浓度(8.2×107atoms · L−1)作为背景值进行比较,切尔诺贝利事故所排放的放射性物质使两千多公里以外的环境129I水平提高了100—1000倍。结合事故初期测定的部分样品中131I的含量,获得了三处采样点的129I/131I原子比值平均值为21(衰变校正到事故发生时间)(Paul et al,1987)。基于这一比值,通过分析切尔诺贝利事故高污染区域土壤剖面和人体甲状腺样品的129I,可以重建事故发生后短时间内未及时测量地区的131I水平,研究131I对人类辐射危害水平与甲状腺癌发病率的关系(Kutschera et al,1988;Hou et al,2003;Michel et al,2005)。
2011年3月的日本福岛核事故也向环境释放了包括131I和129I在内的大量放射性核素。Hou et al(2013)根据测得的福岛附近海水剖面中129I以及其它放射性核素的水平,估算福岛核事故向环境共排放了大约1.2 kg的129I (Hou et al,2013)。此次核事故不但向大气释放了大量放射性核素,同时也向海洋排放了大量放射性物质,不仅导致日本当地环境(土壤、海水、降水、地下水)中的129I水平升高(Miyake et al,2012;Hou et al,2013;Tumey et al,2013;Xu et al,2013,2016b),也使邻近海水中129I和其他放射性核素水平明显提高,截止到2015年,其向海洋排放的放射性物质已扩散至美国西海岸(Stan-Sion et al,2015)。通过对福岛核电站西北60 km处的福岛大学收集的时间序列降水样品中129I进行分析,Xu et al(2013)发现129I浓度在核事故发生后升高了4个数量级,并测得其中129I/131I原子比为16±1,这个值与在福岛核电厂周边土壤中测得的129I/131I比值(22.3 ± 6.3)基本一致,也与切尔诺贝利事故排放的129I/131I原子比(19±5) 接近(Kutschera et al,1988)。利用这一比值并通过测定福岛事故高污染地区土壤中129I水平,Muramatsu et al(2014)准确重建了日本福岛事故高污染区环境131I水平。Xu et al(2016a)之后发现事故后一年内福岛降水中129I水平呈指数衰减,接近事故前129I水平后呈现出季节性变化,推测与沉降于地表的129I向大气中释放/再悬浮有关(Xu et al,2016a)。Piñero-García and Ferro-García(2012)根据西班牙环境样品(雨水、大气、植被等)中放射性核素数据并结合后向和前向气团轨迹分析,探讨了福岛核事故后放射性云团的运动轨迹:先从福岛扩散到太平洋和日本北部,再往北冰洋、加拿大和美国北部运动,然后穿过大西洋,到达西班牙东南部。在加拿大和西班牙的降雨样品及格陵兰岛地区的降雪样品中均检测到了福岛129I的输入,即很好地证实了这一论点(Piñero-García and Ferro-García,2012Herod et al,2015;Gómez-Guzmán et al,2016)。
3.1.2 核燃料后处理厂129I的排放和环境影响
20世纪50年代以来,不少国家和地区建造并运行了核燃料后处理厂,其中部分目前仍在运行。截止到2012年,法国的La Hague核燃料后处理厂已向英吉利海峡海水中排放了约4677 kg的129I,向大气排放了约77 kg的129I;英国的Sellafi eld核燃料后处理厂已向爱尔兰海排放了1634 kg的129I,向大气排放了约197 kg的129I (Daraoui et al,2016),成为目前环境中129I的两大主要来源,使欧洲海域海水中129I/127I比值升高到10−8— 10−4(Hou et al,2001,2007),陆生环境样品(植被、地衣、草等)中129I/127I比值也升高到10−6— 10−4(Duffa and Fréchou,2003;Frechou and Calmet,2003)。另外,德国的WAK、美国的Hanford、日本Tokai和印度核乏燃料后处理厂附近129I/127I比值均显著升高,达10−6— 10−4(Muramatsu et al,1988;Doshi et al,1991;Fritz and Patton,2006)。表2列出了世界上主要核乏燃料后处理厂的累计129I排放量,可以看出除法国的La Hague和英国的Sellafi eld外,法国的Marcoule,美国的Hanford和俄罗斯的Mayak也向环境排放了大量129I (>100 kg) 。除了La Hague和Sellafi eld后处理厂外,其它后处理厂的129I排放主要为大气排放,少量进入河流。
核后处理厂向海洋排放的129I,会通过洋流运动大范围扩散,已观察到La Hague和Sellafi eld后处理厂海洋排放的129I经长距离传输进入北极海域,并进一步扩散至格陵兰海域(He et al,2013)。另外,排放到海水中的129I还会通过挥发性碘进入大气造成二次大气释放,并与直接大气排放的129I一起,在对流层经过较长的滞留(颗粒态碘为14 d,气态无机碘为10 d,气态有机碘为18 d)(Jabbar et al,2012),大范围扩散后沉降到陆地表面。欧洲降水中的129I及其形态分析表明其129I大部分来源于后处理海洋排放的129I从海水的二次释放(Hou et al,2009a;Michel et al,2012)。有研究发现欧洲核后处理厂排放出的129I经过远距离的气态传输,到达并沉降于美国和东亚地区(Moran et al,1999;Toyama et al,2013)。大量研究表明, 欧洲核燃料后处理厂排放的129I已使欧洲、亚洲、北美环境水样中的129I水平均比核活动前提高了1 — 5个数量级(Buraglio et al,2001b;Moran et al,2002;Kekli et al,2003;Aldahan et al,2006;Zhang et al,2011)。
据估计全球核电厂运行已经产生了68000 kg的129I(到2005年),这些129I大部分被封存在核乏燃料元件中(Hou and Hou,2012),目前只有5% — 10%通过乏燃料后处理释放到环境中。随着乏燃料后处理量的增加,环境中的129I水平会继续升高。近年来,我国核能发展迅速,越来越多的核电站投入使用(Ueda et al,2015),我国首座动力堆乏燃料后处理中试厂也已投入使用,且计划建立大容量核燃料后处理厂,这将导致我国环境129I水平的持续上升。研究129I的环境水平和行为,不但对于评价核设施的环境安全非常重要,而且对于评价其长期环境和健康影响非常关键。
3.2 碘的地球化学循环示踪
由于碘主要存在于海洋中,一般认为陆地环境中的碘主要来源于海洋,海洋释放的碘进入大气后,通过扩散、干沉降和湿沉降传输到陆地环境,然后通过径流回到海洋(Fuge and Johnson,1986),图4为碘的地球化学循环的主要路径。129I具有明确的人工来源,与稳定碘的化学性质相同,可用于示踪稳定碘的环境地球化学循环过程(Hou,2004)。利用129I研究碘的地球化学循环主要集中在海洋系统,尤其是海水的水团运输和洋流循环方面(Hou et al,2007;Liu et al,2016),但近年来越来越多的降水及地表水中129I的研究对大气及陆地环境中碘的交换,以及碘在陆地环境的地球化学循环研究提供了极好的研究思路和数据。
研究发现,碘从海洋表面到大气中的释放主要是通过海浪形成的水雾(以I−和形式),以及通过细菌、浮游植物、藻类等将无机碘转化成挥发性的I2和有机碘(如CH3I、CH2I2、CH3BrI、CH2ClI等)的方式完成(Buraglio et al,2001b)。进入大气中的挥发性碘化合物在大气中经过光解等大气化学过程,最终以颗粒态、无机气态和有机气态三种形式存在。研究表明海洋沿岸地区分子碘(I2)含量很高,其主要来源与海藻的释放有关(Kupper et al,2008)。大气中的碘沉降到地表主要是通过湿沉降方式,直接干沉降的贡献很小(Englund et al,2010),129I沉降量与降水量呈明显的正相关(Gómez-Guzmán et al,2012)。降水中碘的形态直接反映大气中碘的存在形式。研究发现,丹麦降水中129I主要形态为碘化物,而127I主要为碘酸盐,这与这两种碘同位素的来源有关。欧洲降水中129I主要来源于核燃料后处理厂海洋排放的129I从海水中的二次释放,欧洲北海、挪威海区域沿岸海水中的129I水平高,通过海藻活动将129I主要以I2的形式释放到大气中,再通过大气化学反应转化为碘化物等其他形态 (Hou et al,2009a)。

图4 碘的地球化学循环主要路径(改自Muramatsu et al(2004),Aldahan et al(2006),徐思琦等(2010))Fig.4 The main pathways of the geochemical cycle of iodine(Modifi ed from Muramatsu et al (2004), Aldahan et al (2006), Xu et al (2010))
陆地环境中碘的循环主要包括大气沉降的碘在土壤中的吸附和迁移、碘从土壤到植物的转移,植物和土壤中碘向大气中的释放,植物和土壤微生物对土壤中碘的转移及其向大气中释放等(Muramatsu et al,2004)。由于土壤中稳定碘的来源复杂,除了大气沉降累积外,还有土壤原基岩中的碘通过风化作用的贡献,因此仅通过分析陆生环境中稳定碘的含量难以定量研究碘的生物地球化学循环。目前环境中129I主要为人为来源,特别是排放源附近环境中129I来源简单,为研究陆生环境中碘的地球化学循环提供独特的示踪剂。Moran et al (2002)通过对美国、加拿大和西欧等地区河流中的127I及129I分析,发现河流129I的通量与该地区大气降水的129I沉降通量接近,表明129I在流域停留时间较短,新进入河流中的129I会很快被带走。采集于全球范围内河水和湖水样品中129I的分析表明南半球中高纬度地区129I/127I比值比赤道和南极地区高,除了与南半球中纬度地区相对较多核武器试验有关外,该地区由于微生物作用将土壤中的碘固定,进入到河流或湖泊中的127I量较少,导致129I/127I比值偏高(Snyder and Fehn,2004)。有研究发现,表层土壤和临近水系中的129I/127I比值接近,这可能归因于河水和湖水中的碘主要来源于表层土壤的浸出 (Rao and Fehn,1999)。另外,大量研究数据表明湖水的129I水平一般低于降水,这与大气中不断升高的129I水平有关。湖水中的129I除了部分来源于大气的直接沉降外,大部分来源于河流注入和周围土壤的淋滤输入,而河流中的129I也主要来源于大气沉降经土壤吸附和淋滤作用后的输入,因此大气沉降的129I经土壤颗粒物吸附,与土壤中原有127I进行交换,淋滤进入河流和湖泊的129I浓度和129I/127I均有所降低 (Reithmeier et al,2007)。以上研究虽然从不同方面提高了对碘在陆地系统的转移的认识,但对碘在大气-降水-地表水-土壤之间循环的深入认识还需要进行更多的研究。
4 总结和展望
本文综述了环境水样中碘及其同位素129I的来源、水平及其在环境中的空间与时间分布。由于碘具有多种形态,包括I−、和有机结合态碘,特别是陆地环境水样中有机碘含量较多,因此在进行样品中129I的含量测定时,必需考虑这部分碘的贡献。高有机碘含量的环境样品中129I分析流程主要包括预处理、有机碘分解、分离富集纯化、靶源制备和129I测量等。本文评述了主要的有机碘分解方法,样品中碘的分离纯化、富集浓缩的方法,129I的各种测量方法以及水样中129I的形态分析方法。
环境水样中的129I主要来源于人类核活动,包括核燃料后处理厂、核武器试验、核事故及核反应堆运行中的泄漏排放。这些核活动向环境中释放了大量的129I,使环境中129I水平显著升高,因此可利用环境水样中129I水平来进行核环境安全监测和评估。不同地区由于受核活动影响的差异,129I水平在全球范围内呈现较大的空间差异。对放射性排放源附近地区(如核燃料后处理厂及核事故发生地周围),可以利用129I监测其环境辐射影响,而对于远离排放源的地区,可以通过分析129I的环境水平作为环境背景值来进行长期影响评价。而且,长时间的核环境监测数据可以形成核活动排放的记录,是评价核排放历史和分析现状的基础。
除了进行核环境安全监测,还可以利用129I进行环境过程示踪研究。通过对大气降水、河水、湖水等环境水样及大气、土壤等不同环境介质中的129I及其形态的研究,可以研究碘在大气-降水-陆地系统中碘的循环过程。但是由于陆地环境中碘含量低,与海水相比,进行淡水中129I的化学形态分析需要很大的样品量才能达到测定要求,且实验过程复杂、耗时长。因此,目前关于环境水样中129I化学形态方面的研究主要集中在海洋系统的洋流循环及水团运动方面,而在陆地系统碘的地球化学循环示踪方面的研究较少。随着129I及其化学形态分析技术的不断完善,尤其是淡水中129I化学形态分析技术的发展,利用129I示踪环境水体中碘的传输和迁移,以及与大气、土壤等介质联合进行陆地碘的生物地球化学循环的研究也会不断深入。
范煜坤. 2013. 我国表土中129I的空间分布及129I年代方法学初探[D].北京: 中国科学院大学. [Fan Y K. 2013. Spatial distribution of129I in Chinese surface soil and preliminary study on the129I chronology [D]. Beijing: University of Chinese Acadamy of Sciences.]
何红蓼, 李 冰, 杨红霞. 2005. 环境样品中痕量元素的化学形态分析: 分析技术在化学形态分析中的应用[J].岩矿测试, 24: 51 – 58. [He H L, Li B, Yang H X. 2005. Trace elemental speciation in environmental samples: analytical techniques applied in chemical speciation [J]. Rock and Mineral Analysis, 24: 51 – 58.]
罗茂益. 2013.129I形态分析及其环境和洋流示踪研究[D].西安: 西安交通大学. [Luo M Y. 2013. Speciation anylysis of129I and its application in environmental and oceanographic trace studies [D]. Xi’an: Xi’an Jiaotong University.]
刘 崴, 杨红霞, 李 冰. 2008. 碘分析方法研究进展[J].岩矿测试, 27: 127 – 136. [Liu W, Yang H X, Li B. 2008. Recent development of methods for iodine analysis [J]. Rock and Mineral Analysis, 27: 127 – 136.]
谢运棉, 班 莹, 蒋崧生,等. 1998. 用串列加速器质谱计测定环境水中129I的浓度[J]. 辐射防护, 19: 81 – 88. [Xie Y M, Ban Y, Jiang S S, et al. 1998. Determination of129I in environmental water using tandem accelerator mass apectrometry [J]. Radiation Protection, 19: 81 – 88.]
徐思琦, 谢周清, 李 冰. 2010. 大气气溶胶中碘形态研究进展[J]. 环境科学, 31: 1121 – 1129. [Xu S Q, Xie Z Q, Li B. 2010. Research progress of atmospheric iodine apeciation in aerosol [J]. Environmental Science, 31: 1121 – 1129.]
张 鸿, 陈应飞, 侯小琳,等. 2012. 深圳大鹏半岛沿岸海水中129I的分布特征和来源[J]. 深圳大学学报(理工版), 29: 1 – 6. [Zhang H, Chen Y F, Hou X L, et al. 2012. The distribution and source apportionment of129I in coastal seawater of Shenzhen Dapeng Peninsula [J]. Journal of Shenzhen University (Science & Engineering), 29: 1 – 6.]
朱发庆, 谭见安. 1988. 我国降水、降尘中硒、碘、氟的研究[J]. 环境科学学报, 8: 428 – 437. [Zhu F Q, Tan J A. 1988. Selenium, iodine and fluorine in rainwater and dustfall in China [J]. Acta Scientiae Circumstantiae, 8: 428 – 437.]
Aldahan A, Alfimov V, Possnert G. 2007a.129I anthropogenic budget: Major sources and sinks [J]. Applied Geochemistry, 22: 606 – 618.
Aldahan A, Kekli A, Possnert G. 2006. Distribution andsources of129I in rivers of the Baltic region [J]. Journal of Environmental Radioactivity, 88: 49 – 73.
Aldahan A, Persson S, Possnert G, et al. 2009. Distribution of127I and129I in precipitation at high European latitudes [J]. Geophysical Research Letters, 36. DOI: 10.1029/2009GL037363.
Aldahan A, Possnert G, Alfi mov V, et al. 2007b. Anthropogenic129I in the Baltic Sea [J]. Nuclear Instruments and Methods in Physics Research Section B, 259: 491 – 495.
Álvarez F, Reich M, Snyder G, et al. 2016. Iodine budget in surface waters from Atacama: Natural and anthropogenic iodine sources revealed by halogen geochemistry and iodine-129 isotopes [J]. Applied Geochemistry, 68: 53 – 63. Bouisset P, Lefevre O, Cagnat X, et al. 1999. Direct gamma-X spectrometry measurement of129I in environmental samples using experimental self-absorption corrections [J]. Nuclear Instruments and Methods in Physics Research Section A, 437: 114 – 127.
Brown C F, Geiszler K N, Lindberg M J. 2007. Analysis of129I in groundwater samples: Direct and quantitative results below the drinking water standard [J]. Applied Geochemistry, 22: 648 – 655.
Buraglio N, Aldahan A, Possnert G. 2000. Analytical techniques and applications of129I in natural water [J]. Nuclear Instruments and Methods in Physics Research Section B, 172: 518 – 523.
Buraglio N, Aldahan A, Possnert G. 2001a.129I in lakes of the Chernobyl fallout region and its environmental implications [J]. Applied Radiation and Isotopes, 55: 715 – 720.
Buraglio N, Aldahan A, Possnert G, et al. 2001b.129I from the nuclear reprocessing facilities traced in precipitation and runoff in northern Europe [J]. Environmental Science & Technology, 35: 1579 – 1586.
Cecil D W, Hall L F, Green J R. 2003. Reevaluation of background iodine-129 concentrations in water from the Snake River Plain aquifer, Idaho, 2003 [R]. Idaho Falls: U.S. Geological Survey Water-Resources Investigations Report 03-4106.
Chen N, Hou X L, Zhou W J, et al. 2014. Analysis of lowlevel129I in brine using accelerator mass spectrometry [J]. Journal of Radioanalytical & Nuclear Chemistry, 299: 1965 – 1971.
Cox R J, Pickford C J. 1992. Determination of Iodine-129 in vegetable samples by inductively coupled plasma mass spectrometry [J]. Journal of Analytical Atomic Spectrometry, 7: 635 – 640.
Dang H, Hou X, Roos P, et al. 2013. Release of iodine from organic matter in natural water by K2S2O8oxidation for129I determination [J]. Analytical Methods, 5: 449 – 456.
Daraoui A, Riebe B, Walther C, et al. 2016. Concentrations of iodine isotopes (129I and127I) and their isotopic ratios in aerosol samples from Northern Germany [J]. Journal of Environmental Radioactivity, 154: 101 – 108.
Doshi G R, Joshi S N, Pillai K C. 1991.129I in soil and grass samples around a nuclear reprocessing plant [J]. Journal of Radioanalytical and Nuclear Chemistry-Letters, 155: 115 – 127.
Duffa C, Fréchou C. 2003. Evidence of long-lived I and Pu isotopes enrichment in vegetation samples around the Marcoule Nuclear Reprocessing Plant (France) [J]. Applied Geochemistry, 18: 1867 – 1873.
Edwarssd R R. 1962. Iodine-129: its occurrence in nature and its utility as a tracer [J]. Science, 137: 851 – 853.
Englund E, Aldahan A, Hou X L, et al. 2010. Iodine (129I
and127I) in aerosols from northern Europe [J]. Nuclear Instruments & Methods in Physics Research B, 268: 1139 – 1141.
(HULQVNLV6SRODRULUFKJHRUJ7HWDO Determination of129I in arctic snow by a novel analytical approach using IC-ICP-SFMS [J]. Journal of Analytical Atomic Spectrometry, 29: 1827 – 1834.
Fabryka-Martin J. 1985. Natural iodine-129 as an environmental tracer [J]. Geochimica et Cosmochimica Acta, 49: 337 – 347.
Fan Y K, Hou X L, Zhou W J. 2013. Progress on129I analysis and its application in environmental and geological researches [J]. Desalination, 321: 32 – 46.
Fehn U, Holdren G R, Elmore D, et al. 1986. Determination of natural and anthropogenic129I in marine-sediments [J]. Geophysical Research Letters, 13: 137 – 139.
Fehn U, Snyder G, Egeberg P K. 2000. Dating of pore waters with129I: relevance for the origin of marine gas hydrates [J]. Science, 289: 2332 – 2335.
Fehn U, Snyder G T, Muramatsu Y. 2007. Iodine as a tracer of organic material:129I results from gas hydrate systems and fore arc fluids [J]. Journal of Geochemical Exploration, 95: 66 – 80.
Frechou C, Calmet D. 2003.129I in the environment of the La Hague nuclear fuel reprocessing plant - from sea to land [J]. Journal of Environmental Radioactivity, 70: 43 – 59.
Fritz B G, Patton G W. 2006. Monitoring iodine-129 in air and milk samples collected near the Hanford Site: an investigation of historical iodine monitoring data [J]. Journal of Environmental Radioactivity, 86: 64 – 77.
Fuge R, Johnson C C. 1986. The geochemistry of iodine - a review [J]. Environmental Geochemistry and Health, 8: 31 – 54.
Gómez-Guzmán J M, Enamorado-Báez S M, Pinto-Gómez A R, et al. 2012. Anthropogenic129I concentration and129I/127I ratio in rainwater from Seville (Spain) in the period 2005—2008 as affected by airborne releases from Sellafield and La Hague facilities [J]. Atmospheric Environment, 56: 26 – 32.
Gómez-Guzmán J M, López-Gutiérrez J M, García-Tenorio R, et al. 2016. Estimating the impact from Fukushima in Southern Spain by131I and accelerator mass spectrometry detection of129I [J]. Journal of Environmental Radioactivity, 166: 36-44.
Gilfedder B S, Lai S C, Petri M, et al. 2008. Iodine speciation in rain, snow and aerosols [J]. Atmospheric Chemistry and Physics, 8: 6069 – 6084.
Gilfedder B S, Petri M, Biester H. 2007a. Iodine and bromine speciation in snow and the effect of orographically induced precipitation [J]. Atmospheric Chemistry and Physics, 7: 2661 – 2669.
Gilfedder B S, Petri M, Biester H. 2007b. Iodine speciation in rain and snow: Implications for the atmospheric iodine sink [J]. Journal of Geophysical Research, 112: 277 – 287.
Gilfedder B S, Petri M, Biester H. 2009. Iodine speciation and cycling in fresh waters: a case study from a humic rich headwater lake (Mummelsee) [J]. Journal of Limnology, 68: 396.
He C, Hou X, Zhao Y, et al. 2011.129I level in seawater near a nuclear power plant determined by accelerator mass spectrometer [J]. Nuclear Instruments and Methods in Physics Research Section A, 632: 152 – 156.
He P, Aldahan A, Possnert G, et al. 2013. A summary of global129I in marine waters [J]. Nuclear Instruments and Methods in Physics Research Section B, 294: 537 – 541.
Herod M N, Clark I D, Kieser W E, et al. 2013.129I dispersion and sources in Northwest Canada [J]. Nuclear Instruments and Methods in Physics Research Section B, 294: 552 – 558.
Herod M N, Suchy M, Cornett R J, et al. 2015. The atmospheric transport of iodine-129 from Fukushima to British Columbia, Canada and its deposition and transport into groundwater [J]. Water Resources Research, 51: 9628 – 9645.
Hou X L. 2004. Application of129I as an environmental tracer [J]. Journal of Radioanalytical and Nuclear Chemistry, 262: 67 – 75.
Hou X L, Aldahan A, Nielsen S P, et al. 2007. Speciation of129I and127I in seawater and implications for sources and transport pathways in the North Sea [J]. Environmental Science & Technology, 41: 5993 – 5999.
Hou X L, Aldahan A, Nielsen S P, et al. 2009a. Time series of129I and127I speciation in precipitation from denmark [J]. Environmental Science & Technology, 43: 6522 – 6528.
Hou X L, Hou Y. 2012. Analysis of129I and its application as environmental tracer [J]. Journal of Analytical Science & Technology, 3: 135 – 153.
Hou X L, Povinec P P, Zhang L, et al. 2013. Iodine-129 in seawater offshore Fukushima: distribution, inorganic speciation, sources, and budget [J]. Environmental Science & Technology, 47: 3091 – 3098.
Hou X L, Dahlgaard H, Rietz B, et al. 1999. Determination of chemical species of iodine in seawater by radiochemical neutron activation analysis combined with ion-exchange preseparation [J]. Analytical Chemistry, 71: 2745 – 2750.
Hou X L, Dahlgaard H, Nielsen S P. 2001. Chemical speciation analysis of129I in seawater and a preliminary investigation to use it as a tracer for geochemical cycle study of stable iodine [J]. Marine Chemistry, 74: 145 – 155.
Hou X L, Fogh C L, Kucera J, et al. 2003. Iodine-129 and Caesium-137 in Chernobyl contaminated soil and their chemical fractionation [J]. Science of the Total Environment, 308: 97 – 109.
Hou X L, Hansen V, Aldahan A, et al. 2009b. A review on speciation of iodine-129 in the environmental and biological samples [J]. Analytica Chimica Acta, 632: 181 – 196.
Hou X L, Zhou W J, Ning C, et al. 2010. Determination of ultralow level129I/127I in natural samples by separation of microgram carrier free iodine and accelerator mass spectrometry detection [J]. Analytical Chemistry, 82:7713 – 7721.
Jabbar T, Steier P, Wallner G, et al. 2012. Iodine isotopes (127I and129I) in aerosols at high altitude Alp stations [J]. Environmental Science & Technology, 46: 8637 – 8644.
Kekli A, Aldahan A, Meili M, et al. 2003.129I in Swedish rivers: distribution and sources [J]. Science of the Total Environment, 309: 161 – 172.
Keogh S M, Aldahan A, Possnert G, et al. 2010. Anthropogenic129I in precipitation and surface waters in Ireland [J]. Nuclear Instruments and Methods in Physics Research Section B, 268: 1232 – 1235.
Krupp G, Aumann D C. 1999. Iodine-129 in rainfall over Germany [J]. Journal of Environmental Radioactivity, 46: 287 – 299.
Kupper F C, Carpenter L J, Mcfi ggans G B, et al. 2008. Iodide accumulation provides kelp with an inorganic antioxidant impacting atmospheric chemistry [J]. Proceedings of the National Academy of Sciences of the United States of America, 105: 6954 – 6958.
Kutschera W, Fink D, Paul M, et al. 1988. Measurement of the129I/131I ratio in Chernobyl fallout [J]. Physica Scripta, 37: 310 – 313.
López-Gutiérrez J M, Garcia-León M, Garcia-Tenorio R, et al. 2000.129I/127I ratios and129I concentrations in a recent sea sediment core and in rainwater from Sevilla (Spain) by AMS [J]. Nuclear Instruments and Methods in Physics Research B, 172: 574 – 578.
López-Gutiérrez J M, Garcia-León M, Schnabel C, et al. 2001. Wet and dry deposition of129I in Seville (Spain) measured by accelerator mass spectrometry [J]. Journal of Environmental Radioactivity, 55: 269 – 282.
López-Gutiérrez J M, Santos F J, Garcia-León M, et al. 2004. Levels and temporal variability of129I concentrations and129I/127I isotopic ratios in atmospheric samples from southern Spain [J]. Nuclear Instruments and Methods in Physics Research Section B, 223 / 224: 495 – 500.
Lehto J, Raty T, Hou X, et al. 2012. Speciation of129I in sea, lake and rain waters [J]. Science of the Total Environment, 419: 60 – 67.
Liu D, Hou X, Du J, et al. 2016.129I and its species in the East China Sea: level, distribution, sources and tracing water masses exchange and movement [J]. Scientifi c Reports, 6: 1 – 9.
Luo M, Hou X, He C, et al. 2013. Speciation analysis of129I in seawater by carrier-free AgI–AgCl coprecipitation and accelerator mass spectrometric measurement [J]. Analytical Chemistry, 85: 3715 – 3722.
Michel R, Daraoui A, Gorny M, et al. 2012. Iodine-129 and iodine-127 in European seawaters and in precipitation from Northern Germany [J]. Science of the Total Environment, 419: 151 – 169.
Michel R, Handl J, Ernst T, et al. 2005. Iodine-129 in soils from Northern Ukraine and the retrospective dosimetry of the iodine-131 exposure after the Chernobyl accident [J]. Science of the Total Environment, 340: 35 – 55.
Miyake Y, Matsuzaki H, Fujiwara T, et al. 2012. Isotopic ratio of radioactive iodine (129I/131I) released from Fukushima Daiichi NPP accident [J]. Geochemical Journal, 46: 327 – 333.
Moran J E, Fehn U, Teng R T D. 1998. Variations in129I/127I ratios in recent marine sediments: evidence for a fossil organic component [J]. Chemical Geology, 152: 193 – 203. Moran J E, Oktay S, Santschi P H, et al. 1999. Atmospheric dispersal of 129-iodine from nuclear fuel reprocessing facilities [J]. Environmental Science & Technology, 33: 2536 – 2542.
Moran J E, Oktay S D, Santschi P H. 2002. Sources of iodine and iodine 129 in rivers [J]. Water Resources Research, 38. DOI: 10.1029/2001WR000622.
Muramatsu Y, Ohmomo Y, Sumiya M. 1988. Determination of iodine-129 and iodine-127 in environmental samples collected in Japan [J]. Journal of Radioanalytical and Nuclear Chemistry, 123: 181 – 189.
Muramatsu Y, Matsuzaki H, Toyama C, et al. 2014. Analysis of129I in the soils of Fukushima Prefecture: preliminary reconstruction of131I deposition related to the accident at Fukushima Daiichi Nuclear Power Plant (FDNPP) [J]. Journal of Environmental Radioactivity, 139: 344 – 350.
Muramatsu Y, Ohmomo Y. 1986. Iodine-129 and iodine-127 in environmental samples collected from Tokaimura/Ibaraki, Japan [J]. Science of the Total Environment, 48: 33 – 43.
Muramatsu Y, Yoshida S, Fehn U, et al. 2004. Studies with natural and anthropogenic iodine isotopes: iodine distribution and cycling in the global environment [J]. Journal of Environmental Radioactivity, 74: 221 – 232.
Negri A E, Ferná ndez Niello J O, Wallner A, et al. 2013.129I dispersion in Argentina: concentrations in fresh and marine water and deposition fluences in Patagonia [J].Environmental Science & Technology, 47: 9693 – 9698.
Oktay S D, Santschi P H, Moran J E, et al. 2001.129I and127I transport in the Mississippi River [J]. Environmental Science & Technology, 35: 4470 – 4476.
Paul M, Fink D, Hollos G, et al. 1987. Measurement of129I concentrations in the environment after the Chernobyl reactor accident [J]. Nuclear Instruments and Methods in Physics Research B, 29: 341 – 345.
Persson S, Aldahan A, Possnert G, et al. 2007.129I Variability in precipitation over Europe [J]. Nuclear Instruments and Methods in Physics Research Section B, 259: 508 – 512.
Piñero-García G F, Ferro-García M A. 2012. Traces of fi ssion products in southeast Spain after the Fukushima nuclear accident [J]. Journal of Environmental Radioactivity, 114: 146 – 151.
Raisbeck G M, Yiou F, Zhou Z Q, et al. 1995.129I from nuclear fuel reprocessing; potential as an oceanographic tracer [J]. Journal of Marine Systems, 6: 561 – 570.
Rao U, Fehn U. 1999. Sources and reservoirs of anthropogenic iodine-129 in western New York [J]. Geochimica et Cosmochimica Acta, 63: 1927 – 1938.
Reifenhauser C, Heumann K G. 1990. Development of a defi nitive method for iodine speciation in aquatic systems [J]. Fresenius Journal of Analytical Chemistry, 336: 559 – 563.
Reithmeier H, Lazarev V, Kubo F, et al. 2005.129I in precipitation using a new TOF system for AMS measurements [J]. Nuclear Instruments and Methods in Physics Research Section B, 239: 273 – 280.
Reithmeier H, Lazarev V, Ruhm W, et al. 2007.129I measurements in lake water for an estimate of regional129I depositions [J]. Science of the Total Environment, 376: 285 – 293.
Reithmeier H, Lazarev V, Ruhm W, et al. 2010. Anthropogenic129I in the atmosphere: overview over major sources, transport processes and deposition pattern [J]. Science of the Total Environment, 408: 5052 – 5064.
Reithmeier H, Lazarev V, Ruhm W, et al. 2006. Estimate of European129I releases supported by129I analysis in an Alpine ice core [J]. Environmental Science & Technology, 40: 5891 – 5896.
Robens E, Aumann D C. 1988. Iodine-129 in the environment of a nuclear fuel reprocessing plant: I.127I and127I contents of soils, food crops and animal products [J]. Journal of Environmental Radioactivity, 7: 159 – 175.
Santschi P H, Schwehr K A. 2004.129I/127I as a new environmental tracer or geochronometer for biogeochemical or hydrodynamic processes in the hydrosphere and geosphere: the central role of organoiodine [J]. Science of the Total Environment, 321: 257 – 271.
Schnabel C, López-Gutiérrez C, Szidat J M, et al. 2001. On the origin of129I in rain water near Zürich [J]. Radiochimica Acta, 89: 815 – 822.
Schwehr K A, Santschi P H. 2003. Sensitive determination of iodine species, including organo-iodine, for freshwater and seawater samples using high performance liquid chromatography and spectrophotometric detection [J]. Analytica Chimica Acta, 482: 59 – 71.
Schwehr K A, Santschi P H, Elmore D. 2005. The dissolved organic iodine species of the isotopic ratio of129I/127I: A novel tool for tracing terrestrial organic carbon in the estuarine surface waters of Galveston Bay, Texas [J]. Limnology & Oceanography Methods, 3: 326 – 337.
Shinohara K. 2004. Measurement and behavior of14C and129I in the environment [J]. Journal of Radioanalytical and Nuclear Chemistry, 260: 265 – 271.
Snyder G, Aldahan A, Possnert G. 2010. Global distribution and long-term fate of anthropogenic129I in marine and surface water reservoirs [J]. Geochemistry, Geophysics, Geosystems, 11. DOI: 10.1029/2009GC002910.
Snyder G, Fehn U. 2004. Global distribution of129I in rivers and lakes: implications for iodine cycling in surface reservoirs [J]. Nuclear Instruments and Methods in Physics Research Section B, 223 / 224: 579 – 586.
Stan-Sion C, Enachescu M, Petre A R. 2015. AMS analyses of I-129 from the Fukushima Daiichi nuclear accident in the Pacific Ocean waters of the Coast La Jolla - San Diego, USA [J]. Environmental Science Process & Impacts, 17: 932 – 938.
Straume T, Marchetti A A, Anspauch L R, et al. 1996. The feasibility of using129I to reconstruct131I deposition from the Chernobyl reactor accident [J]. Health Physics, 71: 733 – 740.
Sutiez J A, Espmtero A G, Rodriguez M. 1996. Radiochemical analysis of129I in radioactive waste streams [J]. Nuclear Instruments and Methods in Physics Research Section A, 369: 407 – 410.
Szidat S, Schmidt A, Handl J, et al. 2000. Iodine-129: Samplepreparation, quality control and analyses of pre-nuclear materials and of natural waters from Lower Saxony, Germany [J]. Nuclear Instruments & Methods in Physics Research Section B, 172: 699 – 710.
Toyama C, Muramatsu Y, Igarashi Y, et al. 2013. Atmospheric fallout of129I in Japan before the Fukushima accident: regional and global contributions (1963—2005) [J]. Environmental Science & Technology, 47: 8383 – 8390.
Toyama C, Muramatsu Y, Uchida Y, et al. 2012. Variations of129I in the atmospheric fallout of Tokyo, Japan: 1963—2003 [J]. Journal of Environmental Radioactivity, 113: 116 – 122.
Tumey S J, Guiderson T P, Brown T A, et al. 2013. Input of129I into the western Pacific Ocean resulting from the Fukushima nuclear event [J]. Journal of Radioanalytical and Nuclear Chemistry, 296: 957 – 962.
Ueda S, Kakiuchi H, Hasegawa H, et al. 2015. Iodine-129 in water samples collected adjacent to a spent nuclear fuel reprocessing plant in Rokkasho, Japan [J]. Journal of Radioanalytical and Nuclear Chemistry, 303: 1211 – 1215. Wagner M J M, Dittrich-Hannen B, Synal H A, et al. 1996. Increase of129I in the environment [J]. Nuclear Instruments and Methods in Physics Research Section B, 113: 490 – 494.
Wong G T F. 1991. The marine geochemistry of iodine [J]. Reviews in Aquatic Sciences, 4: 45 – 73.
Wong G T F, Cheng X H. 1998. Dissolved organic iodine in marine waters: Determination, occurrence and analytical implications [J]. Marine Chemistry, 59: 271 – 281.
Xing S, Hou X, Aldahan A, et al. 2016. Speciation analysis of129I in seawater using coprecipitation and accelerator mass spectrometry and its applications [J]. Journal of Radioanalytical & Nuclear Chemistry: 1 – 9.
Xing S, Hou X, Aldahan A, et al. 2015. Iodine-129 in snow and seawater in the Antarctic: level and source [J]. Environmental Science & Technology, 49: 6691 – 6700.
Xu S, Freeman S P H T, Hou X L, et al. 2013. Iodine isotopes in precipitation: temporal responses to129I emissions from the Fukushima nuclear accident [J]. Environmental Science & Technology, 47: 10851 – 10859.
Xu S, Zhang L Y, Freeman S P H T, et al. 2016a. Iodine isotopes in precipitation: Four-year time series variations before and after 2011 Fukushima nuclear accident [J]. Journal of Environmental Radioactivity, 155: 38 – 45.
Xu S, Zhang L Y, Freeman S P H T, et al. 2016b.129I and137Cs in groundwater in the vicinity of Fukushima Daiichi nuclear power plant [J]. Geochemical Journal, 50: 287 – 291.
Yiou F, Raisbeck G, Imbaud H. 2004. Extraction and AMS measurement of carrier free129I/127I from seawater [J]. Nuclear Instruments & Methods in Physics Research Section B, 223: 412 – 415.
Yoshida S, Muramatsu Y, Katou S, et al. 2007. Determination of the chemical forms of iodine with IC-ICP-MS and its application to environmental samples [J]. Journal of Radioanalytical and Nuclear Chemistry, 273: 211 – 214.
Zhang L, Zhou W, Hou X, et al. 2011. Level and source of129I of environmental samples in Xi’an region, China [J]. Science of the Total Environment, 409: 3780 – 3788.
Zhang L Y, Hou X L. 2013. Speciation analysis of129I and its applications in environmental research [J]. Radiochimica Acta, 101: 525 – 540.
Zhang S, Schwehr K A, Ho Y F, et al. 2010. A novel approach for the simultaneous determination of iodide, iodate and organo-iodide for127I and129I in environmental samples using gas chromatography-mass spectrometry [J]. Environmental Science & Technology, 44: 9042 – 9048.
Zheng J, Yamada M, Yoshida S. 2011. Sensitive iodine speciation in seawater by multi-mode size-exclusion chromatography with sector-fi eld ICP-MS [J]. Journal of Analytical Atomic Spectrometry, 26: 1790 – 1795.
Analysis of129I in environmental water samples and its application on environmental tracing
JIANG Xuhong1,2,3, HOU Xiaolin1,3, CHEN Ning1,3, ZHANG Luyuan1,3
1. State Key Laboratory of Loess and Quaternary Geology, Institute of Earth Environment, Chinese Academy of Sciences, Xi’an 710061, China
2. University of Chinese Academy of Sciences, Beijing 100049, China
3. Shaanxi Key Laboratory of Accelerator Mass Spectrometry Technology and Application, Xi’an AMS Center, Xi’an 710061, China
Background, aim, and scope129I is the only long-lived radioisotope of iodine (with a half-life of 15.7 million years). There are two main sources of129I in the environment, natural and anthropogenic production.129I is naturally produced through the spallation reaction of atmospheric cosmic rays with Xe in the upper atmosphere, the spontaneous fi ssion of238U and thermal neutron-induced fi ssion of235U in the crust, as well as the neutron capture reaction of Te, but this source only accounts for a small part of the129I inventory in the environment. The main sources of129I in the present environment are releases from human nuclear activities, including nuclear weapons testing, nuclear reactor operation, nuclear accidents and nuclear fuel reprocessing plants (NFRPs). Among them, the releases from the NFRPs account for more than 90% of129I in the environment by now. The releases of anthropogenic129I have increased the129I level in the environmental water samples by 3—6 ordersof magnitude compared to the pre-nuclear level, especially in those areas (e.g. Europe, East Asia and North America) close to the point sources such as NFRPs, Fukushima and Chernobyl accident sites. In addition to the significantly spatial variation of129I level, the records in ice cores, long-term atmospheric depositions and sediment cores have demonstrated variation of129I level with time: besides a peak value in 1963, increased level was also observed after 1990, which is corresponding to the history of anthropogenic releases of129I. The iodine concentration in environmental water samples such as atmospheric precipitation, river water, lake water is generally low, and can exist in multi-species. Materials and methods In the environmental water samples, especially in fresh water, organic iodine is an important specie of iodine, which causes a high huge challenge for the accurate determination of ultralow level of129I in this type of samples. In order to determine the total129I and speciation analysis of129I in water samples, organic iodine needs to be converted into inorganic iodine. A typical analytical procedure is presented for determination of129I and its species including sample pretreatment, iodine separation and concentration, target source preparation and129I measurements. Results This paper summarizes and critically reviews various methods for determination of129I and its species in water samples, including decomposition of organic iodine (e.g. UV irradiation, pyrolysis, alkali digestion, ultrasonic alkali digestion, oxidative digestion), chemical separation of iodine and its species from water (e.g. solvent extraction, ion exchange, direct precipitation, co-precipitation and silver adsorption), as well as the measurement techniques for129I (including γ and X-ray spectrometry and liquid scintillation counting method LSC, neutron activation analysis NAA, accelerator mass spectrometry AMS and inductively coupled plasma mass spectrometry ICPMS). Discussion Due to the active properties of iodine, iodine is easy to disperse and migrate in the environment through different media. With the unique sources of anthropogenic129I and its long half-life,129I has been widely applied as an environmental tracer for geochemical processes of iodine. Conclusions By determination of129I and its species in the water samples, the level of129I and its transportation process in the environment can be used to monitor and evaluate environment safety of nuclear facilities. This paper also shows the study progress of129I as environmental tracer on geochemical cycle of stable iodine and nuclear environmental safety. Recommendations and perspectives With the continuous improvement of129I analysis methods, especially the development of129I chemical speciation analysis in freshwater, the129I tracer would be better used to study the transport and migration of iodine in environmental water bodies, our understanding of the bio-geochemical cycle of iodine will be more and more comprehensive. In addition, to carry on long-term nuclear environmental monitoring can form records of nuclear activity emissions and have a guiding role in assessing emissions history and analyzing the status quo.
129I; water samples; organic iodine; speciation analysis; environmental tracing
Date: 2017-03-08; Accepted Date: 2017-04-12
Ministry of Science and Technology of China (2015FY110800); National Natural Science Foundation of China (41271512, 41603125, 11605207)
HOU Xiaolin, E-mail: houxl@ieecas.cn
2017-03-08;录用日期:2017-04-12
科技部基础性工作专项项目(2015FY110800);国家自然科学基金项目(41271512,41603125,11605207)
侯小琳,E-mail: houxl@ieecas.cn
姜旭宏, 侯小琳, 陈 宁, 等. 2017. 环境水样中129I分析及其在环境示踪中的应用[J]. 地球环境学报, 8(3): 203 – 224.
: Jiang X H, Hou X L, Chen N, et al. 2017. Using lithium isotopes traces continental weathering: Progresses and challenges [J]. Journal of Earth Environment, 8(3): 203 – 224.
