石油污染对土壤微生物群落多样性的影响
2017-07-01李晓楼
李晓楼
石油污染对土壤微生物群落多样性的影响
李晓楼
四川职业技术学院,四川遂宁 629000

李晓楼. 石油污染对土壤微生物群落多样性的影响. 生物工程学报, 2017, 33(6): 968–975.Li XL. Influence of oil pollution on soil microbial community diversity. Chin J Biotech, 2017, 33(6): 968–975.
土壤中的微生物主要有细菌、放线菌、真菌三大类群,微生物在石油污染的土壤中发挥着维持生态平衡和生物降解的功能。文中以四川省遂宁市射洪县某废弃油井周围不同程度石油污染土壤为供试土壤,首先对各组供试土壤的基本理化性质进行测定分析;然后采用平板菌落计数法测定了供试土壤中三大类微生物数量的变化,结果表明:相比未被污染的对照土壤,石油污染的土壤中细菌、放线菌、真菌数量均减少,并且土壤中可培养微生物的数量与土壤含水量呈正相关;再采用454焦磷酸测序技术对土壤中的细菌群落多样性及变化进行16S rRNA基因分析。在所有供试的4个土壤样品中,共鉴定出不少于23 982个有效读取序列和6 123种微生物,相比于未被污染的对照土壤,石油污染土壤中细菌的种类更加丰富,主要优势门类为酸杆菌门、放线菌门、拟杆菌门、绿弯菌门、浮霉菌门和变形菌门。但不同土壤样品中优势菌群的群落结构有所差异,石油污染的土壤中,酸杆菌门、放线菌门和变形菌门的数量最多,未被石油污染的土壤中,放线菌门、拟杆菌门和变形菌门的数量最多。
石油污染土壤,454焦磷酸测序,16S rRNA,微生物群落多样性
原油是一种由各种烃类和有机化合物组成的复杂混合物,原油泄露会带来非常严重的环境问题[1-2]。尽管原油工业会带来相应的经济效益,但石油泄露会影响并改变土壤微生物群落结构和生物地球化学循环,同时对土壤肥沃的可持续性和环境质量带来非常严重的负面影响。石油污染物进入土壤后会影响植被的生长和人类的健康[3]。
土壤作为微生物主要的栖息地,是地球环境中生物多样性最丰富的地方[4]。土壤微生物对维持环境生态系统平衡发挥着至关重要的作用。此外,土壤中微生物群落的多样性与其周围的生态系统密切相关,是保持土壤生产力的重要组分之一。近年来,在污染土壤领域有很多关于土壤中微生物的物种多样性[5]、遗传多样性[6]、结构多样性[7]和功能多样性[8]的研究。
随着科学技术的不断进步,现代分子生物学方法能够极大地帮助我们了解微生物群落及其多样性[9]。但是对于一些微生物成分复杂的样品,分子生物学方法只能检测样品中的一些优势菌群,而不能鉴定样品中微生物种类及含量[10-11]。由罗氏公司发明的454焦磷酸测序技术可以对细菌的16S rRNA基因进行焦磷酸测序并分析,从而能够鉴定出样品中细菌的种类及含量。该技术能够克服一般分子生物学方法的局限性,并有效地检测环境中细菌的种群和含量[12-13]。目前该技术已在各种环境中,如废水、生物淤泥及石油污染土壤等样品中检测了细菌群落及多样性[14]。
通过研究石油污染对土壤中的微生物群落影响并评估这些土著微生物降解石油的能力,能够为环境污染的生物修复技术起到强有力的指导作用。本研究首先对各组供试土壤的基本理化性质进行测定分析;然后采用平板计数法测定在不同程度石油污染的土壤中细菌、放线菌及真菌数量的变化;再采用454焦磷酸测序技术测定在不同程度石油污染的土壤中原核微生物群落结构及其多样性,为石油污染土壤的微生物修复提供有效的生物修复策略。
1 材料与方法
1.1 供试土壤、样品处理及理化参数的测定
本次实验以四川省遂宁市射洪县某废弃油井周围不同程度石油污染土壤作为供试土壤。在本研究中,一共采集了4个土壤样品,均来自同一废弃油井地区。其中2个样品分别随机取自靠近废弃油井周围的土壤,另外2个样品分别取自离油井100 m (石油轻度污染土壤组) 处和1 500 m (未被石油污染对照组) 处的土壤。所有土壤样品均采集于离地表层20 cm处,每个样品均分为3等份,一份用来测定土壤中的基本理化组分及石油的含量,一份用来测定土壤中3大类微生物数量,一份用来测定土壤中细菌群落的多样性及结构。
采用烘干法[15-16]测定土壤的含水率,步骤为:称取10 g筛好的土样,装入铝盒称重,打开铝盒盖,于105 ℃干燥8 h,再盖上铝盒盖,称重。以如下公式计算含水率:土壤含水率(%)=(湿土重–烘干土重)/烘干土重×100%[16];采用电位计法[17-18]测定土壤pH;采用凯氏定氮法[19]测定土壤中的全氮含量;采用酸溶-钼锑抗比色法[17]测定土壤中的全磷含量;采用重铬酸钾容量法[17]测定土壤中的有机质含量;采用手持电导率仪测定土壤浸出液的电导率(FiveEasy30,Mettler Toledo,Sweden)[17];采用超声-紫外法[17]测定土壤中石油的含量。表1列出了这些土壤的基本成分及特性。
1.2 土壤中可培养微生物数量的测定
采用选择性培养基及平板菌落计数法对土壤中三大类微生物的数量分别进行测定。分离细菌采用牛肉膏蛋白胨培养基:牛肉膏5 g,蛋白胨10 g,琼脂糖15 g,氯化钠5 g,单蒸水定容至1 000 mL pH 7.2,0.1 MPa,121 ℃灭菌后倒板;分离放线菌采用改良后的高氏I号培养基:KNO31 g,FeSO4·7H2O 0.01 g,K2HPO40.05 g,MgSO4·7H2O 0.5 g,NaCl 0.5 g,琼脂18 g,淀粉20 g (先加入少量水调制成糊状),单蒸水1 000 mL,0.1 MPa,121 ℃灭菌后倒平板。临倒平板时加入重铬酸钾溶液,以抑制细菌和霉菌的生长,同时调节pH 7.2–7.4;分离真菌采用酸性马铃薯琼脂培养基:称取200 g新鲜马铃薯,洗干净后去皮切碎,加入1 000 mL单蒸水煮沸30 min,然后采用纱布过滤,滤液定容至 1 000 mL,再加15 g葡萄糖和15–20 g琼脂,充分溶解后趁热再次纱布过滤,分装三角瓶,0.1 MPa,121 ℃灭菌后倒平板备用。
1.3 细菌16S rRNA基因的提取及PCR扩增
采用E.Z.N.A. Soil DNA Kit (OMEGA,Georgia,GA,USA) 从1 g土壤中提取微生物总基因组DNA,每组样品分别提取3次后合并在一起。细菌的16S rRNA基因大约由1 540个核苷酸组成。在本文中采用的16S rRNA基因扩增的通用引物为:正向序列fD1 (5′-AGA GTTTGATCCTGGCTCAG-3′),反向序列E533R (5′-TTACCGCGGCTGCTGGCAC-3′)[19]。PCR扩增的产物为16S rRNA 基因上1–500 bp的DNA片段。PCR扩增反应的总体积为20 μL,包括 5 ng DNA模板,5 μmol/L引物 (0.8 μL),2.5 mmol/L dNTPs (2 μL),4 μL 5×Fast Pfu缓冲液,和0.4 μL Fast Pfu聚合酶(Applied Biosystems,California,CA,USA)。PCR扩增反应程序为:94 ℃,2 min,1个循环;然后94 ℃,30 s;55 ℃,30;72 ℃,1 min的条件下进行35个循环,最后72 ℃,10 min。PCR产物的分子量为450 bp,采用2%琼脂糖凝胶鉴定。
1.4 细菌16S rRNA基因的焦磷酸测序及数据分析
PCR产物用PCR纯化试剂盒(OMEGA,USA) 进行纯化,采用罗氏454焦磷酸测序仪器(Roche 454 Life Sciences,Branford,CT,USA) 对200 ng纯化后的PCR产物进行测序。所得序列用Mothur[20](v.1.21.1) 软件处理分析。为了改善焦磷酸测序数据的质量以及消除随机测序错误的影响,我们从测序的数据库中删除了一些有缺陷的数据,包括:q平均值小于25,有效读取序列小于200 bp等[21]。然后将所得到的序列与Mothur提供的RDP参考序列和分类系统进行比对和分类。采用基于序列相似度的方法将序列分为不同的操作分类单位OTUs (Operational taxonomic units,即OTUs,序列相似度大于97%可定义为同一种OTU),每一个OTU通常被视为一个微生物物种。相似性小于97%就可以认为属于不同的种,相似性小于93%–95%,可以认为属于不同的属。样品中的微生物多样性和不同微生物的丰度都是基于对OTU的分析。
1.5 数据统计
采用SPSS 17.0统计软件对供试土壤基本成分的数据进行分析统计。采用GraphPaD Prism 5作图软件及-test对供试土壤微生物数量进行作图统计。
2 结果与分析
2.1 供试土壤理化性质研究
2.2 供试土壤中可培养微生物数量的测定
从图1中可以看出,石油污染土壤中含有的细菌、放线菌及真菌的数量相比于未被污染的对照组均有所减少,并且土壤中总的可培养微生物数量与土壤中水分的含量呈现出明显的正相关趋势。另外在未被污染的土壤中放线菌的数量远远大于另外3种土样的数量,表明在被石油污染的土壤中,放线菌的生长受到抑制。
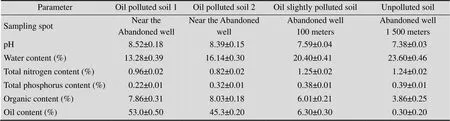
表1 供试土壤的基本理化性质分析
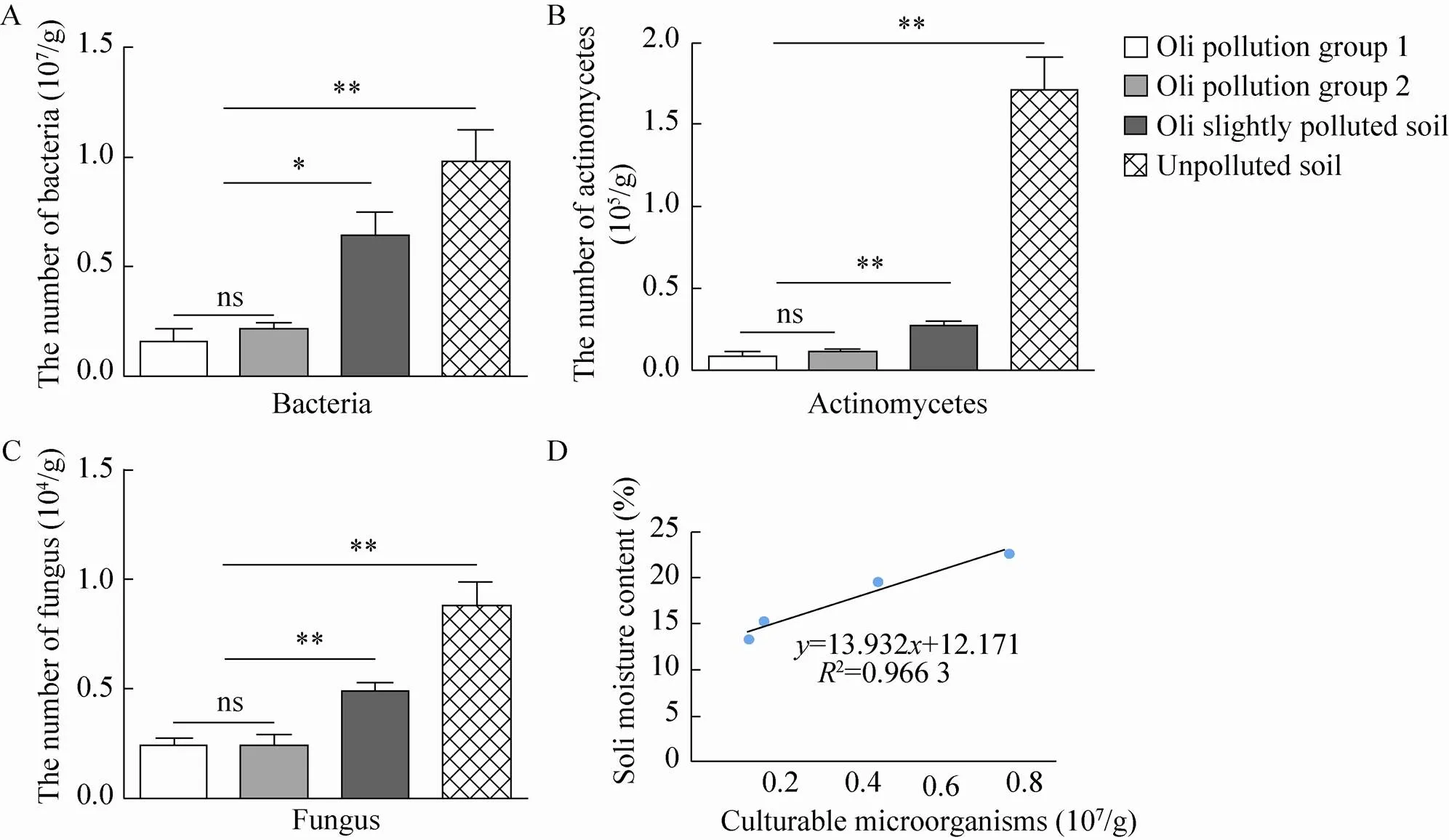
图1 供试土壤中各种微生物的数量与含水量的关系
2.3 供试土壤中细菌群落多样性的研究
表2表示样品经过454焦磷酸测序并分析后的结果,其中从4组样品中一共分析了23 982个有效读取序列。在所有的土壤样品中一共有 6 246个操作分类单位 (Operational taxonomic units,OTUs)。在两组石油污染的土壤中,其OTUs数值均明显高于未被污染的土壤对照组。Chao1丰富度估计量 (Chao1 richness estimator)、香农-威纳多样性指数 (Shannon-Wiener diversity index)、辛普森多样性指数 (Simpson diversity index) 等测量数据均表明被石油污染后的土壤中细菌的种类增加。
2.4 供试土壤中微生物的群落结构分析
根据测序结果,我们一共从4组土壤样品中分析出29个不同的门类。其中,酸杆菌门 (Acidobacteria)、放线菌门 (Actinobacteria)、拟杆菌门 (Bacteroidetes)、绿弯菌门 (Chloroflexi)、浮霉菌门 (Planctomycetes) 和变形菌门(Proteobacteria) 为优势菌群,共占据细菌总含量的80%左右。然而在不同的土壤样品中,优势菌群的群落结构有所差异。从图2中可以看出,在石油污染的土壤中,酸杆菌门 (Acidobacteria)、放线菌门 (Actinobacteria) 和变形菌门 (Proteobacteria) 的含量最多。在未被石油污染的土壤中,放线菌门 (Actinobacteria)、拟杆菌门 (Bacteroidetes) 和变形菌门 (Proteobacteria) 的含量最多。并且,在石油污染的土壤中,变形菌门 (Proteobacteria)、浮霉菌门 (Planctomycetes)、绿弯菌门 (Chloroflexi) 和酸杆菌门 (Acidobacteria) 的含量明显多于未被污染的对照组,而拟杆菌门 (Bacteroidetes)、放线菌门 (Actinobacteria) 的含量则明显少于未被污染对照组。
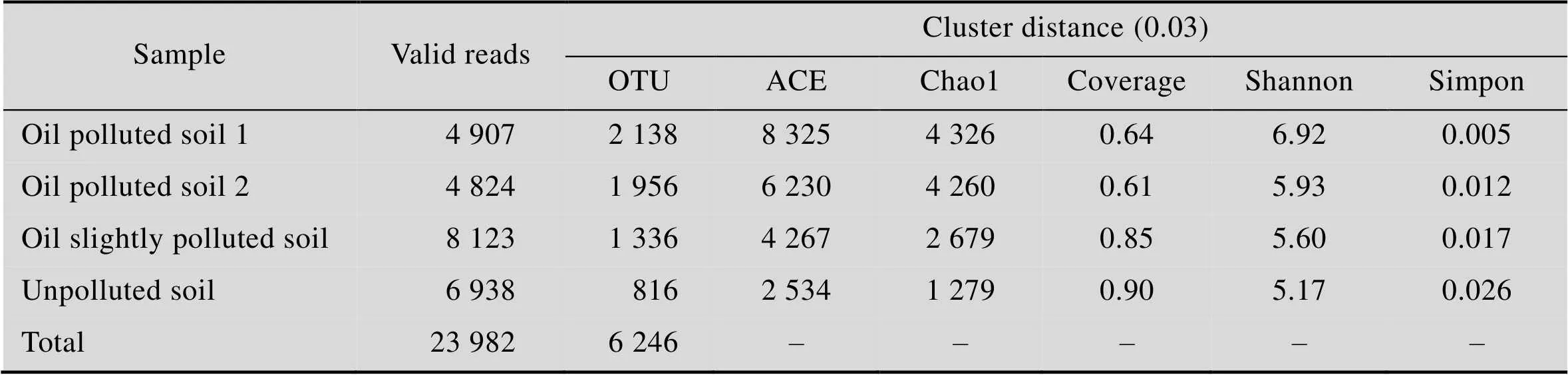
表2 供试土壤样品中细菌群落的多样性
–: Data not available.
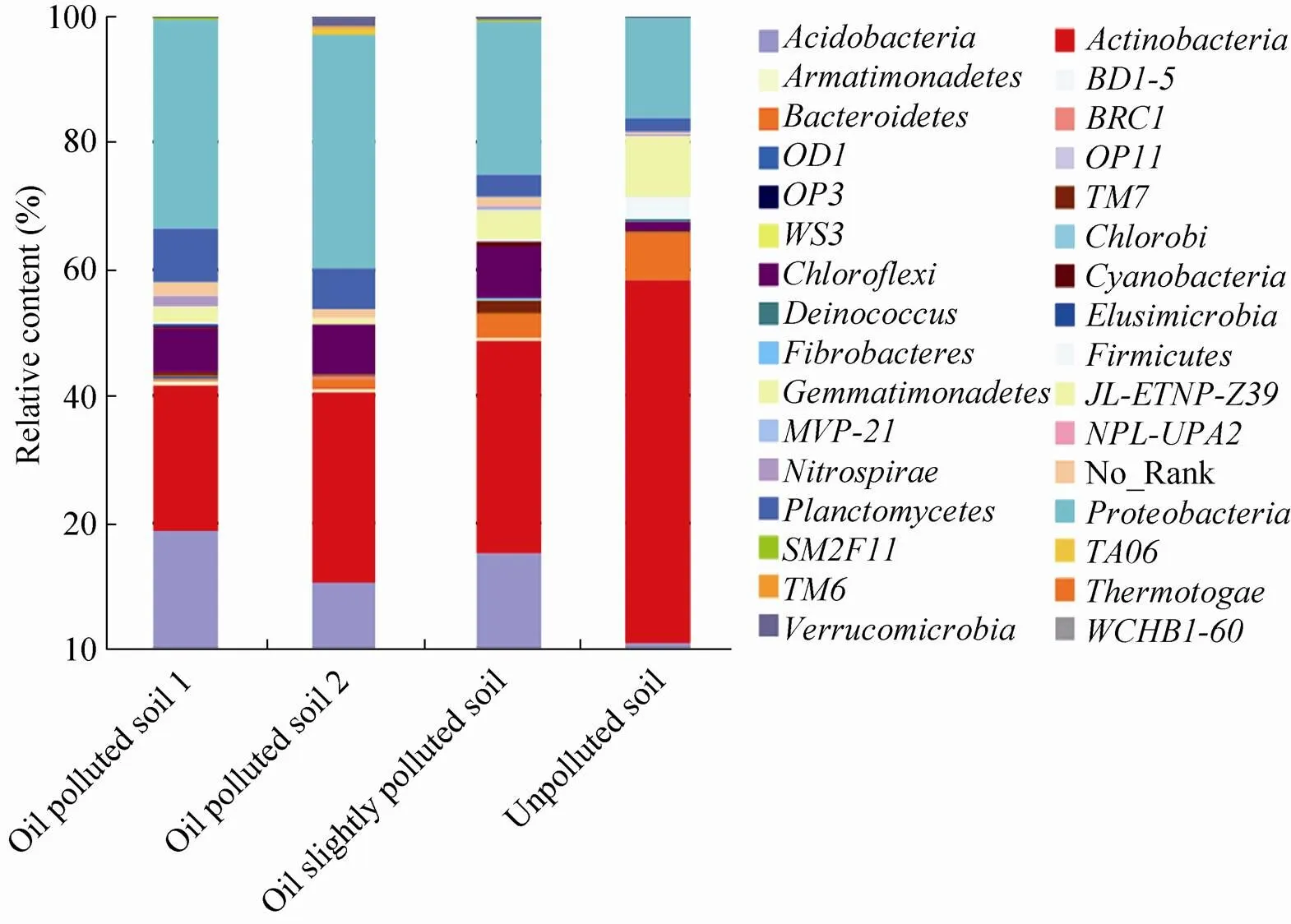
图2 供试土壤样品中主要菌落的相对含量
3 结论与讨论
迄今为止,大量关于石油污染的生物修复研究都集中在从油污土壤中分离出细菌的混合物,然而却很少有人去研究油污土壤中微生物的群落结构及其多样性。在本文中,我们分析了四川省遂宁市射洪县某废弃油井周围油污土壤中各种微生物的群落结构及其多样性,实验结果证明:相比于无油污对照土壤,油污土壤中的微生物种类更加丰富,群落结构更加复杂。
1.1 资料来源 纳入标准:符合国际尿控协会PFD诊断标准[6];单胎妊娠;阴道自然分娩;无会阴侧切;产道轻度撕裂伤;无盆腔感染等病史;子宫脱垂POP-Q 0度、Ⅰ度、Ⅱ度;自愿参与本研究;治疗耐受力和依从性良好。排除标准:泌尿系、生殖系统感染、肿瘤等疾病;产道严重撕裂伤;认知障碍或有精神疾病;治疗依从性差;抵触本研究。分组方案:采用随机数字表法随机分为研究组和对照组,两组患者基线资料对比具有可比性(P>0.05)。见表1。
Sutton等[23]的研究结果表明,在长期受柴油污染的土壤中,其土壤中的微生物群落结构及多样性发生了改变,土壤中的主要优势菌群有变形菌、厚壁菌门、放线菌、酸杆菌门和绿弯菌门。然而,在我们的研究中,如图2所示,石油污染的土壤中主要的优势菌群有酸杆菌门、放线菌门、拟杆菌门、绿弯菌门、浮霉菌门和变形菌门,其中放线菌门和浮霉菌门对石油的降解起到了重要的作用[23]。此外,相比于未被污染的对照土壤组,石油污染的土壤组中的酸杆菌门的含量明显升高,这可能是由于石油污染组的土壤偏碱性导致的,这也与供试土壤测量的pH值结果相符合 (表1)。因此,在不同类型的油污土壤中,其微生物的群落结构和主要优势菌群存在一定的差异。
在本研究中,我们发现土壤中微生物的数量与土壤的含水率表现出一定的正相关趋势,这一结论与任芳菲等[24]的研究结果相符,为干旱地区油污土壤的治理,增加油污土壤中微生物含量,进而提高微生物的降解效率提供依据和思路。油污土壤处于长期湿润和长期干燥两种条件下,土壤中微生物的数量及多样性可能存在差异,由于受条件限制,本文没有深入研究。
本研究报告了石油污染的土壤中微生物多样性的变化,并利用焦磷酸测序技术对石油污染的土壤中原核微生物的群落结构及多样性进行了分析。结果表明,相比于干净的土壤,油污土壤中的微生物种类更加丰富,能够降解油污的菌群明显增多,主要优势菌群发生改变。这些数据能够帮助我们更好地了解和利用各种土著微生物在石油污染土壤微生物修复中的作用。
[1] Zhang ZZ, Gai LX, Hou ZW, et al. Characterization and biotechnological potential of petroleum-degrading bacteria isolated from oil-contaminated soils. Bioresour Technol,2010, 101(21): 8452–8456.
[2] Kato T, Haruki M, Imanaka T, et al. Isolation and characterization of long-chain-alkane degradingfrom deep subterranean petroleum reservoirs. J Biosci Bioeng, 2001, 91(1): 64–70.
[3] Atlas RM. Microbial degradation of petroleum hydrocarbons: an environmental perspective. Microbiol Rev, 1981, 45(1): 180–209.
[4] Tamames J, Abellán JJ, Pignatelli M, et al. Environmental distribution of prokaryotic taxa. BMC Microbiol, 2010, 10(1): 85.
[5] Laureys D, De Vuyst L. Microbial species diversity, community dynamics, and metabolite kinetics of water kefir fermentation. Appl Environ Microbiol, 2014, 80(8): 2564–2572.
[6] Giebler J, Wick LY, Harms H, et al. Evaluating T-RFLP protocols to sensitively analyze the genetic diversity and community changes of soil alkane degrading bacteria. Eur J Soil Biol, 2014, 65: 107–113.
[7] Köchling T, Sanz JL, Gavazza S, et al. Analysis of microbial community structure and composition in leachates from a young landfill by 454 pyrosequencing. Appl Microbiol Biotechnol, 2015, 99(13): 5657–5668.
[8] Trivedi P, He ZL, Van Nostrand JD, et al. Huanglongbing alters the structure and functional diversity of microbial communities associated with citrus rhizosphere. ISME J, 2012, 6(2): 363–383.
[9] Theron J, Cloete TE. Molecular techniques for determining microbial diversity and community structure in natural environments. Crit Rev Microbiol, 2000, 26(1): 37–57.
[10] Ye L, Zhang T. Bacterial communities in different sections of a municipal wastewater treatment plant revealed by 16S rDNA 454 pyrosequencing. Appl Microbiol Biotechnol, 2013, 97(6): 2681–2690.
[11] Claesson MJ, O’Sullivan O, Wang Q, et al. Comparative analysis of pyrosequencing and a phylogenetic microarray for exploring microbial community structures in the human distal intestine. PLoS ONE, 2009, 4(8): e6669.
[12] Bibby K, Viau E, Peccia J. Pyrosequencing of the 16S rRNA gene to reveal bacterial pathogen diversity in biosolids. Water Res, 2010, 44(14): 4252–4260.
[13] Kirchman DL, Cottrell MT, Lovejoy C. The structure of bacterial communities in the western Arctic Ocean as revealed by pyrosequencing of 16S rRNA genes. Environ Microbiol, 2010, 12(5): 1132–1143.
[14] dos Santos HF, Cury JC, do Carmo FL, et al. Mangrove bacterial diversity and the impact of oil contamination revealed by pyrosequencing: bacterial proxies for oil pollution. PLoS ONE, 2011, 6(3): e16943.
[15] Tang J, Wang L, Zhang S. Investigating landscape pattern and its dynamics in Daqing. Int J Remote Sens, 2005, 26(11): 2259–2280.
[16] Scandalios JG. Oxygen stress and superoxide dismutases. Plant Physiol, 1993, 101(1): 7–12.
[17] Lu RK. Soil Argrochemistry Analysis Protocoes. Beijing: China Agriculture Science Press, 1999 (in Chinese).鲁如坤. 土壤农业化学分析方法. 北京: 中国农业科技出版社, 1999.
[18] Blakemore LC, Searle PL, Daly BK. Methods for chemical analysis of soils. New Zealand Soil Bureau Scientific Report 80. Wellington: New Zealand Soil Bureau, 1987: 80.
[19] Marín M, Garcia-Lechuz JM, Alonso P, et al. Role of universal 16S rRNA gene PCR and sequencing in diagnosis of prosthetic joint infection. J Clin Microbiol, 2012, 50(3): 583-589.
[20] Schloss PD, Westcott SL, Ryabin T, et al. Introducing mothur: open-source, platform- independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol, 2009, 75(23): 7537–7541.
[21] Caporaso JG, Lauber CL, Walters WA, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci USA, 2011, 108(S1): 4516–4522.
[22] Jin WB, Li XZ, Long FL, et al. Ecological research on microorganism in crude oil polluted soil. Environ Protect Oil Gas Fields, 1998, 8(3): 2–4 (in Chinese). 金文标, 李秀珍, 龙凤乐, 等. 油污土壤微生物生态研究. 油气田环境保护, 1998, 8(3): 2–4.
[23] Sutton NB, Maphosa F, Morillo JA, et al. Impact of long-term diesel contamination on soil microbial community structure. Appl Environ Microbiol, 2013, 79(2): 619–630.
[24] Ren FF. Studies on physico-chemcal properties and microbial community functional diversity diversity in petroleum contaminationed soil[D]. Harbin: Northeast Forestry University, 2009 (in Chinese). 任芳菲. 石油污染土壤的理化性质和微生物群落功能多样性研究[D]. 哈尔滨: 东北林业大学, 2009.
(本文责编 陈宏宇)
Influence of oil pollution on soil microbial community diversity
Xiaolou Li
Sichuan Vocational and Technological College, Suining 629000, Sichuan, China
Bacteria, actinomycetes and fungi are the three major groups of soil microbes. Soil microbes play a critical role in ecological and biodegradation processes in petroleum-contaminated soils. Based on the actual situation, this study took the oil polluted soil around the abandoned oil well in Shehong County, Suining City, Sichuan Province as the test soil. First, we determined the physiochemical properties of the tested soil; then we analyzed the changes of physiochemical properties and the three major microbes in petroleum contaminated soils. The number of the three major microbes in contaminated soils was relatively fewer than uncontaminated samples, and the water content of the soil was in positive correlation with the number of microbes. Also we assessed the soil bacteria community diversity and changes therein in petroleum-contaminated soils using 454 pyrosequencing of 16S rRNA genes. No less than 23 982 valid reads and 6 123 operational taxonomic units (OTUs) were obtained from all 4 studied samples. OTU richness was relatively higher in contaminated soils than uncontaminated samples. Acidobacteria, Actinobacteria, Bacteroidetes, Chloroflexi, Planctomycetes and Proteobacteria were the dominant phyla among all the soil samples. However, the prokaryotes community abundance of phyla was significantly different in the four samples. The most abundant OTUs associated with petroleum-contaminated soil sample were the sequences related to Acidobacteria, Actinobacteria and Proteobacteria, whereas the most abundance sequences with uncontaminated sample were those related to Actinobacteria, Bacteroidetes and Proteobacteria.
petroleum contaminated soil, 454 pyrosequencing, 16S rRNA, microbial community diversity
10.13345/j.cjb.160467
December 2, 2016; Accepted:February 9, 2017
Xiaolou Li. Tel:+86-825-2221989; E-mail: nh3749@163.com
Supported by:Sichuan Provincial Department of Education Key Projects (No. 13ZA0036).
四川省教育厅重点基金项目(No. 13ZA0036) 资助。
