利用微滴式数字PCR技术分析转基因玉米抗除草剂标记基因EPSP拷贝数
2017-07-01李向龙张中保邹华文吴忠义
王 犇,张 春,李向龙,张中保,邹华文,吴忠义
(1.长江大学 农学院,主要粮食作物产业化湖北省协同创新中心,湖北 荆州 434023;2.北京市农林科学院 北京农业生物技术研究中心,北京市农业基因资源和生物技术重点实验室,北京 100097)
利用微滴式数字PCR技术分析转基因玉米抗除草剂标记基因EPSP拷贝数
王 犇1,2,张 春2,李向龙2,张中保2,邹华文1,吴忠义2
(1.长江大学 农学院,主要粮食作物产业化湖北省协同创新中心,湖北 荆州 434023;2.北京市农林科学院 北京农业生物技术研究中心,北京市农业基因资源和生物技术重点实验室,北京 100097)
基于微滴式数字PCR(Droplet digital PCR,ddPCR)平台,以转基因玉米为例,建立了转基因作物(Genetically modified crops,GM crops)外源基因拷贝数分析方法,对待测样品进行了快速鉴定,并从T0转基因玉米株系中鉴定出多个单拷贝单株。对该方法与实时荧光定量PCR(Quantitative real-time PCR,qRT-PCR)方法在分析结果的准确性方面进行了比较,从试验数据可以看出,2种检测方法的结果比较一致,单拷贝检测结果高度一致;但是ddPCR试验操作更加简便,试验结果可重复性强,试验数据更加准确可靠。研究表明,ddPCR方法是一种更加便捷、快速和准确的外源基因拷贝数分析新方法,基于其在准确性和灵敏度方面的显著优势,将会在转基因作物的外源基因拷贝数分析中得到广泛的应用。
微滴式数字PCR;转基因玉米;外源基因;拷贝数;绝对定量
随着转基因技术的广泛应用,转基因产品检测技术研究的一个关键点在于精准地鉴定转基因植物外源基因拷贝数[1]。外源基因的表达情况与其整合到受体基因组中的拷贝数有密切关系,一般以低拷贝(1~2个)整合时可高效表达,而多拷贝数的整合往往会造成外源基因不稳定表达甚至基因沉默[2]。目前,实时荧光定量PCR(Quantitative real-time PCR,qRT-PCR)、Southern 印迹杂交(Southern Blot)等经典方法已经成熟的应用于转基因生物外源基因拷贝数的分析中。
qRT-PCR技术是一种经典的DNA定量方法,最常用的方法主要有非特异性SYBR Green I染料法和特异性TaqMan探针法2种,其中TaqMan探针法是在定性PCR的基础上添加一条特异性的寡核苷酸荧光探针,其5′端标记一个报告荧光集团,3′端标记一个淬灭荧光集团,两者之间构成能量传递结构。在探针完整时,报告集团发射的荧光信号被淬灭集团吸收,检测系统检测不到荧光信号。PCR扩增时,Taq酶的5′-3′外切酶活性将与底物结合的探针酶切降解,使荧光集团和淬灭集团分离,荧光检测系统可以检测到荧光信号,即每扩增一条DNA链就有一个荧光分子形成,实现了荧光信号的累积与PCR产物形成完全同步,从而实现了定量。TaqMan探针法的成功在于以下2个方面:一是Taq聚合酶所具有的双链特异性的5′-3′外切酶活性;二是利用荧光能量传递技术构建的双标记寡核苷酸探针(TaqMan探针)。实时荧光定量PCR一般使用Ct值(每个反应荧光信号达到设定阈值所需的循环数)定量,Ct值与模板初始拷贝数的对数呈线性关系,利用已知起始拷贝数的标准品可制作标准曲线,只要获得未知样品的Ct值,即可代入方程计算出该样品的起始拷贝数[3]。qRT-PCR方法在分析中需要构建标准曲线,由于利用标准曲线进行定量本身就是一种并不十分精确的相对定量方法,而且建立标准曲线的过程中需要在体系的摸索和优化上投入大量的时间和精力,致使试验周期比较长[4],所以qRT-PCR不是理想的外源基因拷贝数分析方法[5]。传统的Southern Blot方法在分析时存在操作步骤繁琐、样本量大、工作量大、周期长和准确性较差等缺点,并且使用放射性同位素标记会对人体和环境造成较大安全隐患;特别是玉米的基因组较大,杂交信号弱,因此该方法在鉴定T0转基因玉米拷贝数方面难度非常大。
近年来,在核酸定量分析技术领域中,数字PCR(Digital PCR,dPCR)是一种发展迅速的突破性技术。1992年Sykes等[6]尝试在复杂背景下检测低丰度的IgH重链突变基因时,对样品进行有限稀释,使每个反应孔中只存在单个模板分子,通过收集PCR扩增后的信号确定起始分子数,并且确定了以“终点信号的有或无”作为定量标准的数字PCR检测的原则,这也正是数字PCR的雏形。1999年Vogelstein等[7]提出了数字PCR的概念, 将反应体系均匀分配到大量反应单元中,每个反应单元中不包含或包含1个到少数几个目的核酸分子,在每个反应单元中独立地进行PCR扩增,然后逐一检测其荧光信号,再根据泊松分布和荧光信号呈阳性的反应单元数占所有反应单元数的比例来计算目的核酸序列的拷贝数。数字PCR技术具有极高的灵敏度、特异性和精确性,因此被迅速应用于生命科学研究的多个领域,如拷贝数变异分析[8-10]、基因表达分析[11-13]、转基因成分检测[14-16]、基因突变检测[17-20]、微生物检测[21-24]、测序文库质控[25-26]、测序结果验证[27-31]、无创产前诊断[32-34]等。
微滴式数字PCR(Droplet digital PCR,ddPCR)是一种基于泊松分布原理的对待检核酸靶分子实现其浓度或拷贝数鉴定的绝对定量技术,目前在检测核酸分子的绝对计数或定量方面发挥着越来越重要的作用。2015年,中国检验检疫科学研究院朱水芳老师实验室联合国内多家实验室完成了ddPCR用于转基因生物(Genetically modified organisms,GMOs)检测的研究[35]。根据形成反应单元的方式不同,数字PCR主要分为微反应室(孔板)、微流控芯片和微滴等3个系统[36]。ddPCR作为数字PCR技术的一种,包含两部分核心内容:微滴化和微滴分析。微滴化是将配好的包含模板、引物或探针、Mix等成分的20 μL ddPCR预混液制备成20 000个均一的微滴,每个微滴约为1 nL,微滴中或者含有一个至数个待检核酸靶分子,或者不含有靶分子,而且每个微滴均被微滴生成油所包裹并作为一个独立的PCR反应器。区别于qRT-PCR的实时检测,ddPCR是在扩增结束后,在微滴分析仪中逐一对每个微滴的荧光信号进行采集,有荧光信号的说明含有目标靶分子记为阳性微滴(判读为1),没有荧光信号的说明没有目标靶分子则记为阴性微滴(判读为0),最终分析软件统计阳性微滴数占总微滴数的比例,结合泊松分布原理计算出目标靶分子的绝对浓度或拷贝数(拷贝/μL)。由此看出ddPCR的优点是:不依赖于扩增曲线的循环阈值,以分子计数的方式直接获得样本的拷贝数浓度,这样的绝对定量结果便于直接进行横向或纵向比较;更高的精确度,对同一个样本的技术重复的检测能获得一致的结果,变异系数小;更好的重复性,即在不同的实验室之间,或同一实验室不同的检测批次间,能对同一个样本获得一致的定量结果;更高的灵敏度,能更灵敏地检测出其他技术无法检出的或超低丰度的靶标序列;更高的准确度,更准确测定样本中靶标序列的真实含量,真正实现绝对定量分析。
ddPCR作为一种全新的核酸分子定量技术,相较于传统PCR和qRT-PCR,其结果具有更高的准确性、精确度和灵敏度,已被广泛应用于生命科学研究的多个领域,如绝对定量、稀有突变检测和拷贝数变异分析等[36]。本研究采用ddPCR技术开展T0转基因玉米株系中抗除草剂筛选基因EPSP的拷贝数分析。
1 材料和方法
1.1 试验材料
冬季在海南三亚崖城转基因玉米试验基地通过花粉管通道法转化获得玉米种子[37-40],玉米种子播种出苗后长到三叶期喷洒200 mg/L的草甘膦除草剂,经筛选获得的具有除草剂抗性的转基因株系视为T0,以后对转基因株系进行自交扩繁并依次获得T1、T2、T3、T4转基因株系。在实际用于测定的T2和T4样品由不同T0纯化而来。
1.2 ddPCR引物设计
试验内参基因选用玉米醇溶蛋白Zein,经过引物筛选和比较,采用以下具有高度专一性和特异性的引物对进行扩增(表1)。
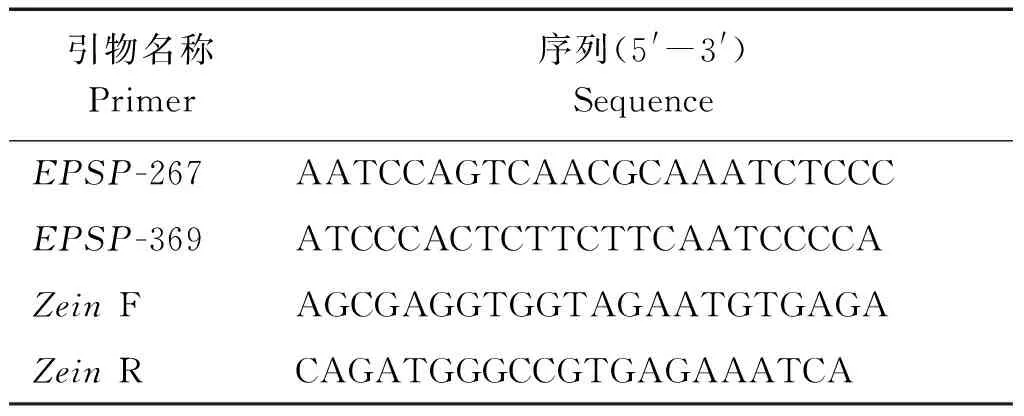
表1 ddPCR引物对序列
1.3 叶片DNA提取
转基因株系叶片基因组DNA的提取采用CTAB法[40-41],浓度测定采用NanoDrop 2000分光光度计检测。为了保证相邻近的2个外源基因插入位点能够被监测到,需要对叶片基因组DNA进行核酸酶的前处理,试验中采用TaKaRa的Hind Ⅲ进行基因组的酶切反应,每微克DNA加入1个单位Hind Ⅲ,在37 ℃条件下水浴处理4 h,65 ℃条件下处理30 min,使酶失活。
1.4 PCR扩增
将模板DNA稀释为20 ng/μL,使用北京农业生物技术研究中心Bio-Rad公司产品QX200ddPCR系统进行ddPCR试验。PCR采用以DNA为模板的EvaGreen染料法,采用20 μL反应体系:2×ddPCR EvaGreen SuperMix 10 μL,上、下游引物各0.4 μL,ddH2O 6.2 μL,DNA 模板 3 μL(20 ng/μL)。PCR反应条件:95 ℃预变性5 min;95 ℃变性30 s,58 ℃退火30 s,72 ℃延伸30 s,共40个循环;4 ℃ 5 min,90 ℃固化5 min(Ramp rate=2.0 ℃/s),立即用于检测(4 ℃保存,不超过2 h)。试验重复1次。
1.5 ddPCR试验操作流程
第1步:微滴生成。将配置好的20 μL样品反应体系加到微滴发生卡(DG8 cartridge)中间一排的8个孔内,注意需缓慢打出液体以避免引入气泡;在微滴发生卡最底下一排8个孔中各加入70 μL微滴生成油(DG oil);盖上胶垫(Gasket),注意两边的小孔都要钩牢;放入微滴生成仪中,一般需2 min即可完成微滴生成;微滴生成于微滴发生卡最上面一排孔内,体积为40 μL,转移至96孔板相应位置孔内,用预热好的PX1热封仪进行封膜。第2步:微滴PCR。在PCR仪上进行PCR扩增,注意升降温速度需低于2.5 ℃/s。第3步:微滴分析。将96孔板放入微滴分析仪中,逐个分析读取微滴荧光信号,判断微滴阳性或阴性。第4步:结果分析。分析软件计算出每个样品中目的序列的拷贝数(拷贝/μL),然后根据内参基因的拷贝数计算得出待检测目的基因的拷贝数。
1.6 qRT-PCR检测转基因株系外源基因拷贝数
该内容委托大北农生物技术公司进行测定,均采用TaqMan探针法。试验重复1次。
2 结果与分析
试验中ddPCR反应进展顺利,所检测每个样品的微滴数均在12 000个以上,而且所有微滴能够分成明显的上下两簇,阈值线以上的点表示能够扩增出目的基因的阳性微滴,阈值线以下的点则代表阴性微滴(图1)。试验利用ddPCR检测T2转基因玉米EPSP基因的绝对拷贝数,先后进行2次独立试验,结果表明,重复性较好,测得数值试验误差为0.43%~4.39%,但不同样品之间内参基因的拷贝数相差较大,如样品108-5测得内参基因为784拷贝,而在108-4样品中测得内参基因绝对拷贝数为1 353(表2)。
由表3可知,ddPCR与TaqMan方法检测T4转基因玉米外源基因拷贝数的结果达到高度一致,表明这2种方法在检测外源基因拷贝数方面的可行性和结果一致性。一般方法对T0转基因植株外源基因拷贝数检测难度较大,而ddPCR技术通过检测到的抗除草剂筛选基因EPSP拷贝数与玉米内参基因Zein拷贝数的比值,可以得出转基因玉米中外源EPSP基因的实际拷贝数。从表4可以看出,ddPCR测定的12个T0转基因玉米植株中有6个EPSP基因拷贝数低于1,其他植株均大于1拷贝,最多达到11拷贝,说明ddPCR方法在对T0转基因植株外源基因拷贝数检测方面有一定的优势。

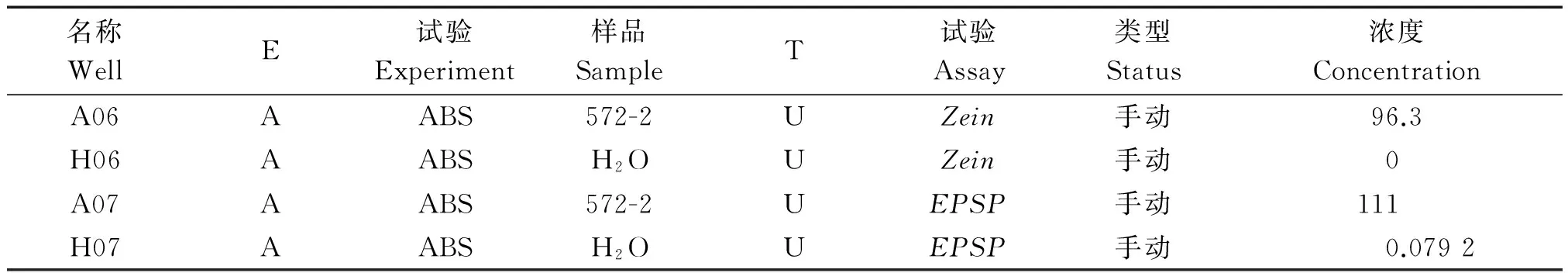
名称WellE试验Experiment样品SampleT试验Assay类型Status浓度ConcentrationA06AABS572-2UZein手动96.3H06AABSH2OUZein手动0A07AABS572-2UEPSP手动111H07AABSH2OUEPSP手动0.0792

A06和A07.572-2样品;H06和H07.水对照。
3 结论与讨论
生物学的基础研究和分子技术的发展伴随着更精确和更灵敏的测量技术的发展。在目前的转基因研究中,能够准确、快速地鉴定转基因生物中外源基因的拷贝数,已经成为转基因产品检测的一个关键内容[42]。除了外源基因的整合拷贝数多少对其是否正常表达至关重要之外,外源基因的整合方式(包括拷贝数、插入位置和侧翼序列等) 也会影响目的基因和蛋白的表达,而且对外源基因在受体中的遗传稳定性具有很大的影响[43]。经典的外源基因拷贝数检测方法主要有qRT-PCR和Southern Blot 2种。其中qRT-PCR 分析方法的步骤较为繁琐,首先需要准备标准DNA样品,然后利用此样品构建标准曲线,而标准曲线的建立又涉及体系的摸索与优化,需要耗费大量的时间和精力,所需试验周期比较长;另外,借助标准曲线进行定量本身就是一种相对定量方法,所以试验结果并不准确。而传统的Southern Blot方法不但对DNA需求量大、试验操作步骤复杂,而且对人员操作的技术要求比较高:转膜必须要充分以保证DNA转到膜上,杂交条件及漂洗要严格控制以保证阳性结果与背景反差对比明显,洗膜要充分但不能过度等;另外,若要用同位素法,对人体健康和环境安全存在严重威胁。此法在分析时的最大问题在于试验工作量大、周期长、结果准确性较差。
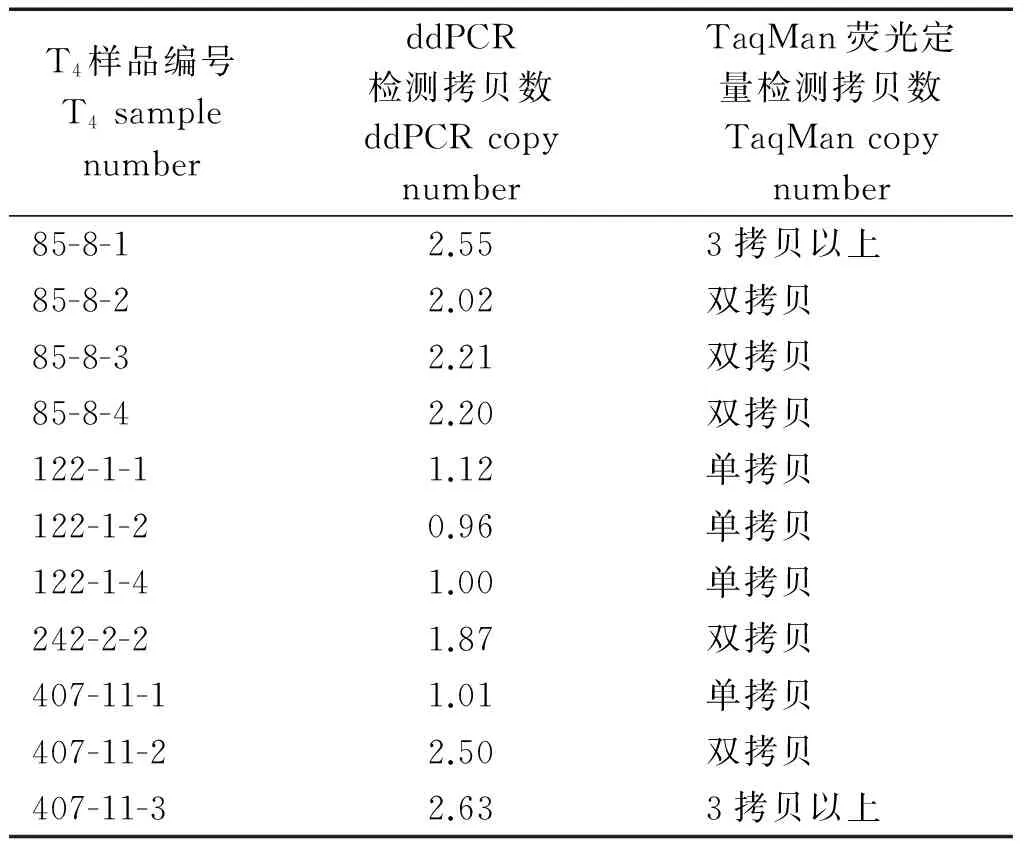
表3 ddPCR与TaqMan检测T4转基因玉米EPSP基因拷贝数结果比较
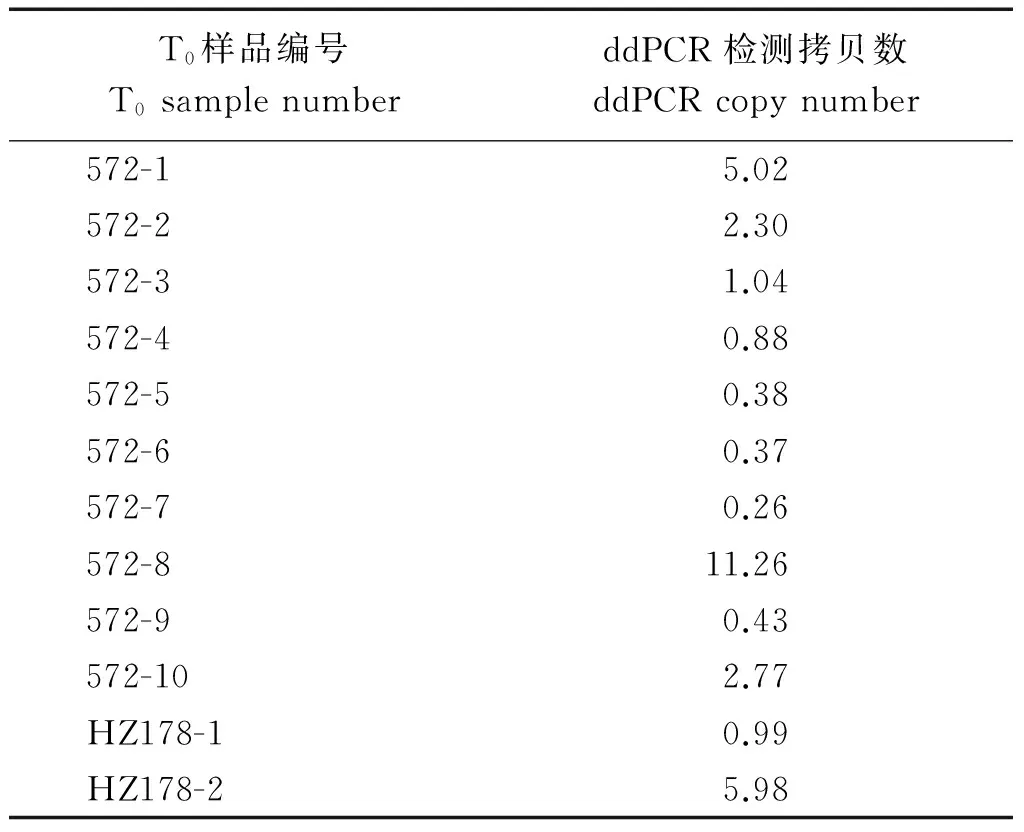
表4 ddPCR测定T0转基因玉米EPSP基因拷贝数
研究通过试验探究了ddPCR方法在分析转基因作物外源基因拷贝数方面的应用。ddPCR作为一种新兴的、准确的绝对定量技术,已经被越来越多的应用于生命科学研究的多个领域,比如核酸绝对定量、稀有突变检测和拷贝数变异等。但目前用ddPCR分析转基因生物的外源基因拷贝数的研究还较少。与传统方法相比较,ddPCR的优势非常明显,主要体现在操作流程更加简单,不需要构建标准曲线,并且是对待检核酸分子直接进行绝对定量,试验结果更加准确可靠。另外,ddPCR在线性范围、检测极限和定量极限等方面都更胜于qRT-PCR[13]。这些优点已得到了其他研究人员的认可[44]。
本研究建立并优化了ddPCR检测试验体系,分析了整合到转基因玉米中的外源基因(EPSP)的拷贝数。结果表明,采用qRT-PCR检测外源基因拷贝数,与ddPCR的检测结果相一致,特别是在对单拷贝转化单株的检测结果上两者高度一致。对比发现,ddPCR检测方法简便,操作简单,不需构建标准曲线,节约时间。结果表明,不同样品之间检测到的内参基因拷贝数相差较大,由于上样DNA总浓度(以NanoDrop光度法计算)是一样的,这可能是因为在浓度测定中无法排除其中杂质的干扰,由此得知,在测定每个试验样品时都需设置内参基因以保证不同样品之间结果的可比性。ddPCR检测T0转基因玉米EPSP基因拷贝数时,存在拷贝数低于0.5的检测单株,可能是目的基因在T0转基因玉米植株中以嵌合体形式存在;其中拷贝数大于1的单株居多,说明通过花粉管通道转化法获得的转基因玉米株系中以多拷贝整合的几率较大;通过该方法能获得一些以理想的拷贝数(单拷贝或双拷贝)整合的转基因玉米株系,以提供试验所需的材料。试验初步建立了T0转基因玉米植株的外源基因拷贝数鉴定方法,能够快速、规模化地鉴定转基因玉米外源基因的拷贝数。
ddPCR方法在试验中充分体现了它的优点:操作简便、节约时间和检测结果准确可重复等,但也存在以下缺点,一是对于检测结果的数值进行拷贝数界定依据不够明确,以双拷贝为例,可以将检测数值为1.80~2.20的单株鉴定为双拷贝,但是对于数值为1.50~1.80或2.20~2.50的单株的鉴定依据并不明确,试验中,编号407-11-2和85-8-1单株的ddPCR检测结果分别为2.50和2.55,而荧光定量检测结果也是分别简单定性为双拷贝和三拷贝及以上,2个样品的ddPCR检测结果很相近,但在荧光定量的检测结果上却相差很大;二是该试验所需试剂和耗材只能使用该仪器生产商匹配的,并且全部依靠进口,因此,存在订货周期长、试剂耗材费用高等不利因素。三是测定的绝对拷贝数是针对引物之间的扩增片段而言,并不代表全长的外源DNA所有信息,要想知道全长的信息,有必要针对DNA不同部位开展ddPCR分析。
利用ddPCR进行拷贝数检测时,待检测样品在反应室中随机、独立分布是单分子成功扩增和准确定量核酸靶分子拷贝数的关键因素,所以研究在设计试验时选用了内切酶Hind Ⅲ对基因组DNA处理。研究是以转基因玉米单株中的筛选标记基因为检测对象,在一定程度上也可以作为目的基因的拷贝数参考;另外可尝试在目的基因的不同位置设计引物进行检测,以保证目的基因拷贝数检测结果的准确性。由于该方法可以对经过除草剂筛选获得的T0单株进行拷贝数鉴定,这是本试验所提到的其他2种方法无法实现的,所以能够最大程度的减少其通过多代自交达到纯合所需的时间,提高选择效率。通过试验发现:ddPCR能够有效节省试验试剂和样品量、对核酸纯度要求不高,试验操作简便省时,具有更高的检测灵敏度和结果准确度,是一种快速、灵敏、精确的转基因作物外源基因拷贝数鉴定新方法。
[1] Heyries K A,Tropini C,Vaninsberghe M,et al.Megapixel digital PCR[J].Nature Methods,2011,8(8):649-651.
[2] 万 艳,李丽玲,陈小佳.应用实时荧光定量PCR检测外源基因拷贝数的新方法[J].暨南大学学报:自然科学与医学版,2009,30(3):310-313.
[3] 王爱民.实时荧光定量PCR(TaqMan)法测定外源基因的拷贝数[J].广西植物,2009,29(3):408-412.
[4] Whale A S,Huggett J F,Cowen S,et al.Comparison of microfluidic digital PCR and conventional quantitative PCR for measuring copy number variation[J].Nucleic Acids Research,2012,40(11):e82.
[5] 姜 羽,胡佳莹,杨立桃.利用微滴数字PCR分析转基因生物外源基因拷贝数[J].农业生物技术学报,2014,22(10):1298-1305.
[6] Sykes P J,Neoh S H,Brisco M J,et al.Quantitation of targets for PCR by use of limiting dilution[J].Bio Techniques,1992,13(3):444-449.
[7] Vogelstein B,Kinzler K W.Digital PCR[J].Proceedings of the National Academy of Sciences of the United States of America,1999,96(16):9236-9241.
[8] Abyzov A,Mariani J,Palejev D,et al.Somatic copy number mosaicism in human skin revealed by induced pluripotent stem cells[J].Nature,2012,492(7429):438-442.
[9] Boettger L M,Handsaker R E,Zody M C,et al.Structural haplotypes and recent evolution of the human 17q21.31 region[J].Nature Genetics,2012,44(8):881-885.
[10] Hindson B J,Ness K D,Masquelier D A,et al.High-throughput droplet digital PCR system for absolute quantitation of DNA copy number[J].Analytical Chemistry,2011,83(22):8604-8610.
[11] Nair V D,Ge Yongchao,Balasubramaniyan N,et al.Involvement of histone demethylase LSD1 in short-time-scale gene expression changes during cell cycle progression in embryonic stem cells[J].Molecular and Cellular Biology,2012,32(23):4861-4876.
[12] Gorbachev A Y,Fisunov G Y,Izraelson M,et al.DNA repair inMycoplasmagallisepticum[J].BMC Genomics,2013,14(1):726.
[13] Jiang Ke,Ren Chunyan,Nair V D.MicroRNA-137 represses Klf4 and Tbx3 during differentiation of mouse embryonic stem cells[J].Stem Cell Research,2013,11(3):1299-1313.
[14] Morisset D,tebih D,Milavec M,et al.Quantitative analysis of food and feed samples with droplet digital PCR[J].PLoS One,2013,8(5):e62583.
[15] Corbisier P,Bhat S,Partis L,et al.Absolute quantification of genetically modified MON810 maize (ZeamaysL.) by digital polymerase chain reaction[J].Analytical and Bioanalytical Chemistry,2010,396(6):2143-2150.
[16] Burns M J,Burrell A M,Foy C A.The applicability of digital PCR for the assessment of detection limits in GMO analysis[J].European Food Research and Technology,2010,231(3):353-362.
[17] Shlush L I,Zandi S,Mitchell A,et al.Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia[J].Nature,2014,506(7488):328.
[18] Johnson B E,Mazor T,Hong Chibo,et al.Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma[J].Science,2014,343(6167):189-193.
[19] Miyaoka Y,Chan A H,Judge L M,et al.Isolation of single-base genome-edited human iPS cells without antibiotic selection[J].Nature Methods,2014,11(3):291-293.
[20] Abdel-wahab O,Klimek V M,Gaskell A A,et al.Efficacy of intermittent combined RAF and MEK inhibition in a patient with concurrent BRAF- and NRAS-mutant malignancies[J].Cancer Discovery,2014,4(5):538-545.
[22] Rothrock M J,Hiett K L,Kiepper B H,et al.Quantification of zoonotic bacterial pathogens within commercial poultry pro-cessing water samples using droplet digital PCR[J].Advances in Applied Microbiology,2013,3(5):403-411.
[23] Zhao Hui,Wilkins K,Damon I K,et al.Specific qPCR assays for the detection ofOrfvirus,PseudocowpoxvirusandBovinepapularstomatitisvirus[J].Journal of Virological Methods,2013,194(1/2):229-234.
[24] Kelley K,Cosman A,Belgrader P,et al.Detection of methicillin-resistantStaphylococcusaureusby a duplex droplet digital PCR assay[J].Journal of Clinical Microbiology,2013,51(7):2033-2039.
[25] Laurie M T,Bertout J A,Taylor S D,et al.Simultaneous digital quantification and fluorescence-based size characterization of massively parallel sequencing libraries[J].BioTechniques,2013,55(2):61-67.
[26] White R A,Grassa C J,Suttle C A.Draft genome sequence of exiguobacterium pavilionensis strain RW-2,with wide thermal,salinity,and pH tolerance,isolated from modern freshwater microbialites[J].Genome Announcements,2013,1(4):e00513-e00597.
[27] Cangi M G,Biavasco R,Cavalli G,et al.BRAFV600E-mutation is invariably present and associated to oncogene-induced senescence in Erdheim-Chester disease[J].Annals of the Rheumatic Diseases,2015,74(8):1596-1602.
[28] Chong I Y,Cunningham D,Barber L J,et al.The genomic landscape of oesophagogastric junctional adenocarcinoma[J].The Journal of Pathology,2013,231(3):301-310.
[29] Frésard L,Leroux S,Servin B,et al.Transcriptome-wide investigation of genomic imprinting in chicken[J].Nucleic Acids Research,2014,42(6):3768-3782.
[30] Wang I X,Core L J,Kwak H,et al.RNA-DNA differences are generated in human cells within seconds after RNA exits polymerase Ⅱ[J].Cell Reports,2014,6(5):906-915.
[31] Chen Rui,Mias G I,Li-pook-than J,et al.Personal omics profiling reveals dynamic molecular and medical phenotypes[J].Cell,2012,148(6):1293-1307.
[32] Holmberg R C,Gindlesperger A,Stokes T,et al.Akonni TruTip(®) and qiagen(®) methods for extraction of fetal circulating DNA-evaluation by real-time and digital PCR[J].PLoS One,2013,8(8):e73068.
[33] Gu Wei,Koh W,Blumenfeld Y J,et al.Noninvasive prenatal diagnosis in a fetus at risk for methylmalonic acidemia[J].Genetics in Medicine,2014,16(7):564-567.
[34] Pornprasert S,Prasing W.Detection of alpha(0)-thalassemia South-East Asian-type deletion by droplet digital PCR[J].European Journal of Haematology,2014,92(3):244-248.
[35] Fu Wei,Zhu Pengyu,Wang C,et al.A highly sensitive and specific method for the screening detection of genetically modified organisms based on digital PCR without pretreatment[J].Scientific Reports,2015,5(1):12715.
[36] Sedlak R H,Jerome K R.Viral diagnostics in the era of digital polymerase chain reaction[J].Diagnostic Microbiology and Infectious Disease,2013,75(1):1-4.
[37] 李志亮,吴忠义,杨 清,等.花粉管通道法在玉米基因工程改良中的应用[J].玉米科学,2010,18(4):71-73,76.
[38] 王艳杰,申家恒.花粉管通道法转基因技术的细胞胚胎学机理探讨[J].西北植物学报,2006,26(3):628-634.
[39] 王秀君,郎志宏,陆 伟,等.利用花粉管通道法将耐草甘膦基因mG2-epsps导入玉米自交系的研究初报[J].中国农业科技导报,2008,10(4):56-62.
[40] 王永锋,栾雨时,高晓蓉.花粉管通道法在植物转基因中的研究与应用[J].东北农业大学学报,2004,35(6):764-768.
[41] 陈向明.用CTAB法提取植物DNA的技术改进[J].合肥教育学院学报,2000(4):14-16.
[42] Zhang Dabing,Guo Jinchao.The development and standardization of testing methods for genetically modified organisms and their derived products[J].Journal of Integrative Plant Biology,2011,53(7):539-551.
[43] Vaucheret H,Béclin C,Elmayan T,et al.Transgene-induced gene silencing in plants[J].The Plant Journal,1998,16(6):651-659.
[44] Hayden R T,Gu Z,Ingersoll J,et al.Comparison of droplet digital PCR to Real-time PCR for quantitative detection of cytomegalovirus[J].Journal of Clinical Microbiology,2013,51(2):540-546.
Analysis of the Copy Number of Herbicide Resistant Marker GeneEPSPin Transgenic Maize by Droplet Digital PCR
WANG Ben1,2,ZHANG Chun2,LI Xianglong2,ZHANG Zhongbao2,ZOU Huawen1,WU Zhongyi2
(1.College of Agriculture,Yangtze University,Hubei Collaborative Innovation Center for Grain Industry,Jingzhou 434023,China;2.Beijing Agro-Biotechnology Research Center,Beijing Academy of Agriculture and Forestry Sciences,Beijing Key Laboratory of Agricultural Gene Resources and Biotechnology,Beijing 100097,China)
The droplet digital PCR (ddPCR) method to evaluate the exogenous gene copy number in genetically modified crops (GM crops) as the example from genetically modified maize was developed,and some T0transgenic maize plants with single copy ofEPSPgene were selected from all the samples to be detected.The results were also compared with those from quantitative real-time PCR (qRT- PCR),as could be seen from the experimental datas,the results from qRT-PCR(TaqMan) and ddPCR forEPSPwere consistent,especially the single copy detection results of both in a high degree of consistency.And the ddPCR method was easy to operate,the result was repeatable and accurate.Therefore,the ddPCR would be well developed as a novel method for estimating transgenic copy number with high accuracy,which might be widely used in the exogenous genes copy number analysis in GM crops in the near future.
Droplet digital PCR; Genetically modified maize; Exogenous gene; Copy number; Absolute quantification
2017-03-10
国家转基因重大专项重点课题(2014ZX0800303B);北京市农林科学院科研能力创新项目;北京市科委项目(Z171100001517001)
王 犇(1988-),男,安徽宿州人,在读硕士,主要从事玉米抗旱分子生物学研究。王犇、张春为同等贡献作者。
邹华文(1973-),男,江苏邳州人,教授,博士,硕士生导师,主要从事植物抗逆分子生物学研究。 吴忠义(1969-),男,福建德化人,研究员,博士,硕士生导师,主要从事植物抗逆分子生物学研究。
S513.03
A
1000-7091(2017)03-0070-07
10.7668/hbnxb.2017.03.011
