磁性氮掺杂石墨烯固相萃取环境水样中的4种有机氯污染物
2017-06-21陈林吉朱晁乐曹小吉周婷叶学敏莫
陈林吉+朱晁乐+曹小吉+周婷+叶学敏+莫卫民
摘 要 采用化学共沉淀法成功合成了磁性氮掺杂石墨烯纳米材料, 对其吸附性能进行了初步探讨。此磁性纳米材料对对氯间二甲苯酚的吸附不局限于均匀的单分子层吸附,吸附动力学符合准二级动力学模型。将其作为磁性固相吸附剂,通过对吸附剂用量、超声萃取时间、水样pH值、上样体积等条件的优化,建立了超声辅助磁性固相萃取气相色谱/串联质谱同时测定环境水样中的三氯生(TCS)、对氯间二甲苯酚(PCMX)、六氯苯(HCB)和2,2′,4,4′,5,5′六氯联苯(PCB153) 4种有机氯污染物的方法。 在优化条件下,将6.0 mg Fe3O4/NG分散于100 mL水样中,调节水样至pH 5,超声萃取15 s,磁性分离,3 mL乙醇和2 mL二氯甲烷分步洗脱,洗脱液氮吹定容,进行气相色譜质谱联用分析。4种有机污染物在0.1~10 μg/L范围内与峰面积呈良好的线性关系,相关系数为0.9983~0.9999,检出限(S/N=3)和定量限(S/N=10)分别为0.05~0.6 ng/L和0.4~2.4 ng/L,3个加标浓度水平的回收率为68.3%~103.4%,日内、日间测定的相对标准偏差分别为3.3%~6.9%和3.4%~9.4%(n=6)。本方法简单方便,易于操作,适用于环境水样中有机氯污染物的检测。
关键词 氮掺杂石墨烯;磁性固相萃取;环境水样;抗菌剂;有机氯污染物
1 引 言
三氯生(Triclosan, TCS)和对氯间二甲苯酚(Parachlormetaxylenol,PCMX)作为广谱性抗菌剂。已被添加在个人护理用品(如肥皂、洗面奶、洗手液、洗发水、牙膏及玩具)中长达数十年[1]。TCS可在分子和细胞水平上对生物造成影响,产生酶和基因毒性,导致生物体组织器官的突变和癌变[2]。2016年9月,美国食品药品监督管理局宣布禁止在洗手液和沐浴露中添加三氯生。六氯苯(Hexachlorobenzene, HCB)和多氯联苯类化合物(PCBs)是典型的有机氯污染物,具有较强的生殖毒性[3],早在30多年前就被禁止使用[4]。我国GB38382002《地表水环境质量标准》[5]和 GB 57492006 《生活饮用水卫生标准》[6]规定我国地表水中HCB的检测限量标准为0.05 mg/L,生活饮用水中HCB的检测限量标准为0.001 mg/L, 多氯联苯总量的检测限量值为0.0005 mg/L。
TCS、HCB以及PCBs等有机氯污染物在环境水样中含量低,再加上复杂基体的干扰,导致其在进仪器分析前,必须进行富集和净化。采用液液萃取[7,8]、固相萃取[9]、液液微萃取[10]等前处理技术富集和净化环境水样中的有机氯污染物,存在操作繁琐耗时、消耗大量有毒有机溶剂以及需要专用设备等问题。基于磁性石墨烯的磁性固相萃取(Magnetic solid phase extraction, MSPE)技术结合了磁性萃取操作简便、无需专业设备和石墨烯具有超大的比表面积和大π共轭体系、对有机化合物具有超大吸附容量的特性,在环境水样中有机氯污染物的前处理方面取得了一定的进展[11,12]。然而,石墨烯作为一种疏水性的碳基纳米材料,在水中容易团聚,限制了其在水样前处理技术中的应用和发展[13]。近年来,科研工作者在石墨烯的化学改性和应用方面也开展了不少工作[14,15]。通过化学掺杂N原子,可以改善石墨烯的亲水性,提高阴极氧化还原反应活性和稳定性[16]。目前,磁性氮掺杂石墨烯(Fe3O4/nitrogendoped graphene, Fe3O4/NG)作为磁性固相萃取剂的研究还未见报道。
本研究采用化学共沉淀法合成了Fe3O4/NG纳米材料,考察了其吸附性能。并将其作为磁性固相吸附剂,在超声辅助下,通过对超声萃取时间、水样pH值、洗脱剂的种类和用量、水样体积等磁性固相萃取条件的系统优化,结合气相色谱/串联质谱(Gas chromatography coupled to tandem mass spectrometry, GCMS/MS)技术,建立一种便捷快速、绿色环保的筛查环境水样中痕量TCS、对氯间二甲苯酚(Parachlometaxylenol, PCMX)、HCB和2,2′,4,4′,5,5′六氯联苯(2,2',4,4',5,5'Hexachlorobiphenyl, PCB153)4种污染物的分析方法。
2 实验部分
2.1 仪器与试剂
Agilent 7890A7000B气相色谱三重四极杆质谱仪(美国 Agilent公司);CTC多功能自动进样器(瑞士 CTC公司);Waters 2695高效液相色谱仪(美国Waters公司),配备Waters 2996型二极管阵列检测器;X′Pert Pro X射线衍射仪(荷兰帕纳科公司);J3426多功能振动样品磁强计(英国Cryogenic公司);Hitachi S4700扫描电子显微镜(日本日立公司);EDAXII X射线能谱仪(美国EDAX公司)。
PCMX、TCS、HCB和PCB153(纯度>99.0%,百灵威公司);氮掺杂石墨烯(阿拉丁试剂(上海)有限公司);甲醇(色谱纯,德国Merck公司);乙醇和正己烷(色谱纯,百灵威公司);二氯甲烷(农残级,百灵威公司);乙酸乙酯(美国Tedia公司);丙酮(色谱纯,江苏永华化学科技有限公司);HCl、NaOH、NaCl、(NH4)2Fe(SO4)2·6H2O、NH4Fe(SO4)2·12H2O(分析纯,杭州华东医药集团公司);N50铷铁硼磁铁(NdFeB,宁波市鄞州冠能磁业有限公司);实验用水为超纯水(美国MilliQ公司)。
2.2 标准溶液的配制
准确称取适量标准品,HCB用正己烷,其余用甲醇配制成1.00 mg/mL 标准储备液,4℃避光保存,使用时,以甲醇稀释至所需浓度。
2.3 气相色谱质谱条件
气相色谱条件:色谱柱:DB5MS(30 m × 0.25 mm i.d. × 0.25 μm);程序升温:100℃保持2 min,以 20℃/min升至300℃;载气He,流速 1 mL/min;进样口温度:300℃;进样量:1 μL;不分流进样。
质谱条件:电子轰击电离源(EI),电离能量70 eV;离子源温度230℃,色谱质谱传输杆温度250℃;质量扫描范围50~500 amu;碰撞气N2:流速1.5 mL/min,淬灭气He:2.25 mL/min;检测模式:多反应监测模式(MRM)。
2.4 Fe3O4/NG的制备和吸附实验
根据文献[17]报道的化学共沉淀法制备Fe3O4/NG。室温下,称取10 mg Fe3O4/NG置于10 mL水样中,加入不同初始浓度(0.1、0.5、1.0、5.0、10和50 mg/L) 的目标物,超声萃取20 s后,采用外加磁场对吸附剂进行分离,取上清液进行HPLCPDA分析。
2.5 磁性固相萃取实验
将10 mL 100 μg/L模拟水样置于40 mL玻璃瓶中,调节水样至pH 5,加入6.0 mg Fe3O4/NG, 超声分散萃取15 s。将磁铁静置于瓶壁外15 s,分离回收Fe3O4/NG。用3 mL乙醇和2 mL二氯甲烷分步超声洗脱Fe3O4/NG上吸附的目标物,磁性分离Fe3O4/NG,合并洗脱液,氮吹至0.8 mL,用乙醇二氯甲烷(1∶1, V/V)溶液定容至1 mL,过0.22 μm尼龙滤膜,待GCMS/MS分析。
3 结果与讨论
3.1 Fe3O4/NG的表征
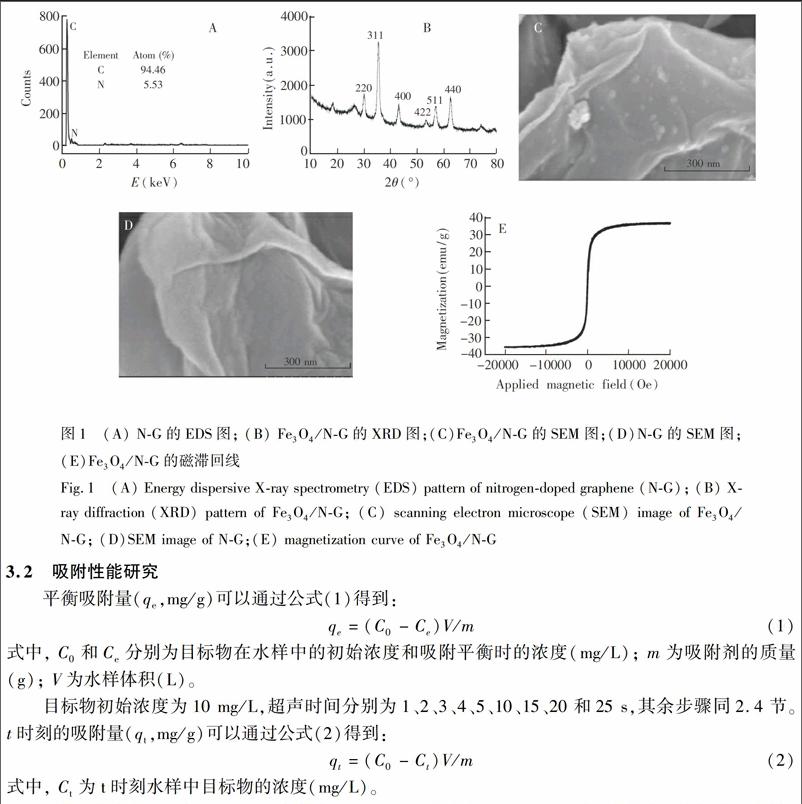
氮掺杂石墨烯的X射线能谱(Energy dispersive Xray spectroscopy,EDS)分析结果如图1A所示,此纳米材料的氮掺杂量为5.53%(w/w)。
图1B为Fe3O4/NG的X射线衍射(Xray diffraction, XRD)图谱,2θ=30.2°、35.6°、43.3°、53.6°、57.2°、62.8°处的衍射峰分别对应纯立方尖晶石晶系Fe3O4的(220)、(311)、(400)、(422)、(511)和(440)晶面(JCPDS卡, 030653107)的特征吸收峰,表明Fe3O4纳米颗粒已成功嫁接到NG上[18]。对比Fe3O4/NG和NG的扫描电镜(Scanning electron microscope, SEM)图1C和图1D,可以确证尺寸为10~20 nm的Fe3O4颗粒零星分布在NG的褶皱层表面[19]。此外,应用振动样品磁强计(Vibrating sample magnetometer, VSM)绘制了Fe3O4/NG的磁滞回曲线。如图1E所示,Fe3O4/NG没有剩磁和磁矫顽力,具有良好的超顺磁性,其饱和磁化强度高达36.93 emu/g,完全满足磁性分离的磁响应要求[20]。
3.2 吸附性能研究
平衡吸附量(qe,mg/g)可以通过公式(1)得到:
式中, C0和Ce分别为目标物在水样中的初始浓度和吸附平衡时的浓度(mg/L); m为吸附剂的质量(g); V为水样体积(L)。
目标物初始浓度为10 mg/L,超声时间分别为1、2、3、4、5、10、15、20和25 s,其余步骤同2.4节。t时刻的吸附量(qt,mg/g)可以通过公式(2)得到:
式中, Ct为t时刻水样中目标物的浓度(mg/L)。
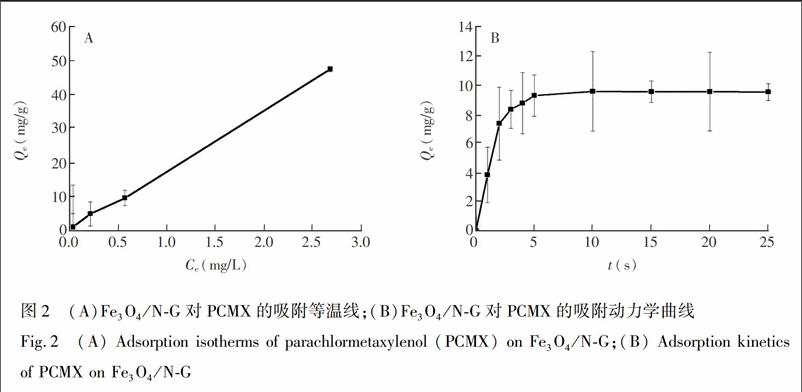
吸附结果如图2A所示,10 mL水样中PCMX的初始浓度为50 mg/L时,吸附等温线依然呈线性。采用Freundlich和Langmuir吸附等温模型拟合PCMX在Fe3O4/NG上的吸附等温线[21],拟合方程的相关系数rF、rL分别为0.9996和0.9995,均大于0.95,说明PCMX在Fe3O4/NG的吸附不局限于均匀的単分子层吸附,还可能存在不均匀的多分子层吸附[22]。由Langmuir吸附等温线可知,298 K时,Fe3O4/NG对PCMX的最大吸附量为238 mg/g。
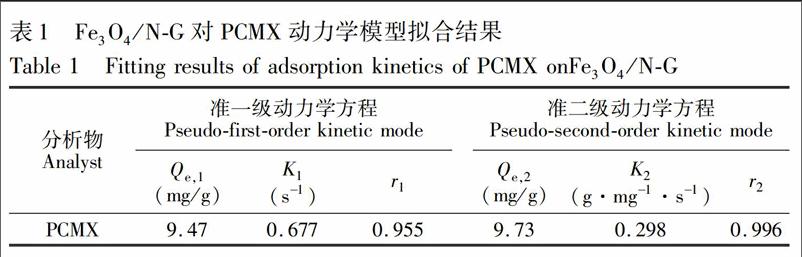
如图2B所示,在0~3 s之间,Fe3O4/NG对PCMX的吸附量随吸附时间延长而迅速增加;3~5 s,吸附量缓慢增加;10 s时,吸附达到平衡,平衡吸附量高达9.58 mg/g。应用准一级动力学和准二级动力学拟合Fe3O4/NG对PCMX的吸附动力学过程[21],结果如表1所示,准二级动力学方程的相关系数r2大于准一级动力学方程的相关系数r1,表明Fe3O4/NG对PCMX的吸附动力学过程符合准二级吸附动力学模型[23]。
由吸附等温实验结果可知,10 mg Fe3O4/NG在20 s内即可完全吸附10 mL水样中50 mg/L的TCS、HCB和PCB153,吸附量达到50 mg/g。
3.3 萃取条件的优化
本实验采用单因素法对影响MSPE萃取回收率的相关条件进行了优化。优化过程中模拟水样中化合物的含量均為1.0 μg。
3.3.1 萃取剂用量的影响 考察了Fe3O4/NG用量(3.0~15.0 mg)对萃取回收率的影响,结果如图3A所示,当Fe3O4/NG用量为6.0 mg时,萃取回收率达到最大值。过多的Fe3O4/NG可能会对4种分析物形成永久性吸附,从而降低萃取回收率。因而,本实验选择的Fe3O4/NG用量为6.0 mg。
3.3.2 超声萃取时间的影响 超声有助于Fe3O4/NG在水中的分散,加大了接触面积,促进了质量传递。如图3B所示,由于掺杂了氮元素,Fe3O4/NG的亲水性明显优于磁性石墨烯[15],仅需超声15 s,萃取即达到平衡。增加超声时间,萃取回收率下降,可能由于过长的超声时间使Fe3O4从氮掺杂石墨烯上脱落下来,导致磁性萃取剂不能完全回收。因而,本实验选取的超声萃取时间为15 s。
3.3.3 水样pH值的影响 水样pH值会影响分析物在水中的存在形式。本实验考察了水样pH=3~11条件下的Fe3O4/NG对HCB、PCB153、TCS和PCMX的萃取回收率,结果如图3C所示,当水样pH=5时,HCB、PCB153、TCS和PCMX的回收率达到最大。当水样pH=3时,部分Fe3O4水解,使得一部分吸附了目标物的氮掺杂石墨烯无法磁性回收,萃取回收率下降。中性物质HCB和PCB153在整个pH范围内保持电中性,当pH>5时,与Fe3O4/NG之间的ππ作用力和疏水作用力不受影响,萃取回收率基本保持不变;另一方面,两种弱酸性物质TCS和PCMX随着pH值的增大,电离加剧,与Fe3O4/NG之间的疏水作用力减弱,萃取回收率降低。最终,确定水样的pH=5。
3.3.4 离子强度和洗脱剂种类及用量的影响 本实验考察了离子强度(0~4% (w/V) NaCl)对萃取回收率的影响,结果表明,不加盐时,萃取回收率最大。因此,随后的实验中都没有加盐。此外,考察了甲醇、乙醇、丙酮、二氯甲烷、正己烷和乙酸乙酯等洗脱剂的洗脱效果,实验结果表明,先用乙醇, 再用二氯甲烷, 洗脱效果较好。随后考察了乙醇和二氯甲烷用量对萃取回收率的影响。结果表明,先用3 mL乙醇洗脱一次, 再用2 mL二氯甲烷洗脱一次, 即可达到满意的效果。
3.3.5 最大上样体积 有机氯污染物在环境水样中的实际残留量是非常低的,加大水样前处理的上样体积,增加富集量,是提高实际检测浓度的有效手段之一。考察了水样体积对回收率的影响,结果如图3D所示,水样体积小于100 mL时,4种目标物的回收率保持在70.0%~105.1%之间,而当水样体积超过100 mL时,萃取回收率明显下降,可能原因是水体积增大,仅用15 s的超声萃取时间,萃取材料无法充分吸附目标物,从而导致萃取回收率降低。本方法最大上样体积为100 mL。
3.4 方法评价
在优化的实验条件下,对4种目标物的线性范围、相关系数、最低检出限及精密度等进行了考察,结果见表2。PCMX、HCB、TCS和PCB153在0.1~10 μg/L浓度范围内与色谱峰面积呈良好的线性关系,其相关系数R2在0.9983~0.9999之间。检出限(S/N=3)和定量限(S/N=10)分别为0.05~0.6 ng/L和0.4~2.4 ng/L,满足国家标准的要求[5~7]。
为了考察本方法的重现性,分别测定了浓度为1 μg/L的水样的日内及日间精密度。日内精密度是通过1天之内平行测定6次水样样品,得到的相对标准偏差。日间精密度是通过连续6天测定同一组水样样品,每天测定一次,得到的相对标准偏差。如表2所示,日内及日间精密度分别是3.3%~6.9%和3.4%~9.4%。
将本方法与文献报道的固相萃取[9]、固相微萃取[24]、磁力搅拌吸附萃取[25]和液液微萃取[10]等方法进行比较,本研究提出的Fe3O4/NG只需15 s即可完成磁性萃取,萃取时间明显缩短,简化了操作过程。
3.5 样品分析
在最佳实验条件下,用建立的Fe3O4/NGMSPE/GCMS/MS方法对本地区的3个环境水样(生活废水、实验室自来水和钱塘江水样)进行分析检测,在生活废水样品中检测到TCS和PCMX两种有机氯污染物,浓度分别为7.21和5.38 μg/L,HCB和PCB153未检出。生活废水和加标浓度为1 μg/L的生活废水的多反应监测(MRM)离子流图见图4。同时,考察了3个加标水平的回收率,结果如表3所示,4种目标物的回收率在68.3%~103.4%之间,可以满足环境水样分析的需求。
4 结 论
采用化学共沉淀法合成了Fe3O4/NG纳米材料,考察了其对4种有机氯污染物PCMX、HCB、TCS和PCB153的吸附性能。结果表明,Fe3O4/NG对4种污染物不仅吸附快,而且有较大的吸附容量;以其为磁性吸附剂,成功建立了同时测定环境水样中4种有机污染物的超声辅助磁性固相萃取氣相色谱串联质谱分析方法。在优化条件下,对于100 mL水样,本方法仅需6.0 mg吸附材料、超声萃取15 s、5 mL洗脱溶剂、15 s的磁分离即可完成磁性萃取过程,操作简单,有机溶剂消耗少,可应用于环境样品中个人护理品类抗菌剂和有机氯污染物的快速筛查。
References
1 Crinnion W J. Altern. Med. Rev., 2010, 15(2): 101-109
2 Guo Q, Yan J, Wen J J, Hu Y Y, Chen Y B, Wu W J. Sci. Total. Environ., 2016, 571: 1304-1311
3 Shao D D, Sheng G D, Chen C L, Wang X K, Nagatsub M. Chemosphere, 2010, 79 (7): 679-685
4 Liu P, Zhang D J, Zhan J H. J. Phys. Chem. A, 2010, 114(50): 13122-13128
5 GB 38382002, Environmental Quality Standards for Surface Water. National Standards of the People′s Republic of China
地表水环境质量标准. 中华人民共和国国家标准. GB38382002
6 GB 57492006, Standards for Dringing Water Quality. National Standards of the People′s Republic of China
生活饮用水卫生标准. 中华人民共和国国家标准. GB 57492006
7 HJ 7152014, Water QualityDetermination of Polychorinated Biphenyls(PCBs)Gas Chromatography Mass Spectrometry. The State Environmental Protection Standard of the People's Republic of China
水質多氯联苯的测定气相色谱质谱法. 中华人民共和国国家环境保护标准. HJ 7152014
8 SHAO Yang, YANG GuoSheng, LIU WeiHua, MA LingLing, LUO Min, HAN Shen, XU DianDou. Chinese J. Anal. Chem., 2016, 44(5): 698-706
邵 阳, 杨国胜, 刘韦华, 马玲玲, 罗 敏, 韩 深, 徐殿斗. 分析化学, 2016, 44(5): 698-706
9 Han Q, Wang Z H, Xia J F, Xia L H, Chen S, Zhang X Q, Ding M Y. J. Sep. Sci., 2013, 36(2122): 3586-3591
10 Chen M J, Liu Y T, Lin C W, Ponnusamy V K, Jen J F. Anal. Chim. Acta, 2013, 767(5): 81-87
11 Luo Y B, Li X, Jiang X Y, Cai B D, Zhu F P, Zhang H F, Chen Z G, Pang Y Q, Feng Y Q. J. Chromatogr. A, 2015, 1406: 1-9
12 Sun T, Yang J, Li L J, Wang X Z, Li X W, Jin Y R. Chromatographia, 2016, 79(5): 345-353
13 Wang X, Liu B, Lu Q P, Qu Q S. J. Chromatogr. A, 2014, 1362(44): 1-15
14 Xiao R, Zhang X T, Zhang X N,Niu J H, Lu M H, Liu X H, Cai Z W. Talanta, 2017, 166: 262-267
15 Menezes H C, de Barcelos S M R, Macedo D F D, Purceno A D, Machado B F, Teixeira A P C, Lago R M, Serp P, Cardeal Z L. Anal. Chim. Acta, 2015, 873: 51-56
16 PENG San, GUO HuiLin, KANG XiaoFeng. Acta Phys. Chim. Sci., 2014, 30(9): 1778-1786
彭 三, 郭慧林, 亢晓峰. 物理化学学报, 2014, 30(9): 1778-1786
17 Qu S, Wang J, Kong J L, Yang P Y, Chen G. Talanta, 2007, 71(3): 1096-1102
18 Cao X J,Shen L X, Ye X M, Zhang F F, Chen J Y, Mo W M. Analyst, 2014, 139(8): 1938-1944
19 Wang W N, Li Y P, Wu Q H, Wang C, Zang X H, Wang Z, Anal. Methods, 2012, 4(3): 766-772
20 Ma Z Y, Guan Y P, Liu H Z. J. Polym. Sci. A, 2005, 43(15): 3433-3439
21 LIU WanYi, YANG LuZe, YU Meng, LIU Miao. Chinese J. Anal. Chem., 2016, 44(5): 707-715
刘宛宜, 杨璐泽, 于 萌, 刘 淼. 分析化学, 2016, 44(5): 707-715
22 OrozcoGuareo E, SantiagoGutiérrez F, MornQuiroz J L, MoranQuiroz, J L, HernandezOlmos S L, Soto V, de la Cruz W, Manriquez R, GomezSalazar S. J. Colloid Interface Sci., 2010, 349(2): 583-593
23 Wang R Z, Huang D L, Liu Y G,Peng Z W, Zeng G M, Lai C, Xu P, Huang C, Zhang C, Gong X M. RSC Adv., 2016, 6(108): 106201-106210
24 Ke Y Y, Zhu F, Zeng F, Luan T G, Su C Y, Ou Yang G F. J. Chromatogr. A, 2013, 1300(14): 187-192
25 PintadoHerrera M G, GonzálezMazo E, LaraMartín P A. Chemosphere, 2014, 95(1): 478-485
