羧基甜菜碱型亲水作用色谱柱的制备及性能研究
2017-06-21常宇阳戴小军龚波林
常宇阳+戴小军+龚波林
摘 要 以甲基丙烯酰氧乙基二甲基乙酸铵(CBMA)为功能单体,利用表面引发原子转移自由基聚合(Surfaceinitiated atom transfer radical polymerization, SIATRP)技术,将CBMA接枝到硅胶表面,得到接枝聚合物CBMA的亲水作用色谱固定相(SilicaCBMA)。通过改变SIATRP反应体系中单体的浓度,制备了3种不同接枝量的亲水作用色谱固定相。考察了SilicaCBMA固定相对有机酸类化合物的分离性能以及流动相中pH值、盐浓度、水含量等因素对溶质保留行为的影响。结果表明,在亲水作用色谱模式下,SilicaCBMA固定相对有机酸类化合物的分离是离子交换作用与亲水作用的混合色谱模式。流动相中盐浓度增大,溶质保留减弱,符合离子交换作用特征; 固定相和溶质的离子化程度受流动相pH值影响较大,pH值增大,溶质保留增强; 而溶质的保留时间随流动相水含量增加而降低则是典型的亲水作用色谱特征。使用自制SilicaCBMA柱,建立了芦丁片中维生素C、芦丁含量的亲水作用色谱测定方法,操作方法简单,为强极性样品的分离测定提供了新方法。
关键词 亲水作用色谱; 固定相; 羧基甜菜碱; 原子转移自由基聚合
1 引 言
亲水作用色谱(Hydrophilic interaction liquid chromatography,HILIC)作为一种分离强极性化合物的液相色谱模式,最早由Alpert于1990年提出[1],其主要特征是使用极性固定相和水/水溶性有机溶剂(通常是乙腈)做流动相。作为HILIC分离的核心,HILIC固定相功能基的结构直接影响其对溶质的选择性和分离效率。常见HILIC固定相可分为两类,一类为小分子键合相,功能基为二醇基、氰基、氨基、酰胺基、半胱氨酸基、麦芽糖基[2]等的固定相; 另一类为聚合物键合相,功能基为聚琥珀酰亚胺[3],聚磷酰胆碱[4]和聚磺酸基甜菜碱[5]的固定相。
含两性甜菜碱型配基的固定相,功能基同时含有两种相反电荷,结构新颖,在20世纪80年代作为两性离子固定相进入色谱领域[6]。在HILIC领域,普遍应用的是含硫代甜菜碱功能基和含磷酰胆碱功能基两性离子的商品柱,在蛋白组学、药物分析、糖类化合物的分离中得到了广泛应用[7~9]。据报道,在溶液pH值3.0~8.0范围内,这2种两性离子功能基表面改性的色谱固定相的ζ电势均为负值,相当于色谱固定相表面被负离子修饰而带负电荷[10,11]。相比较而言,在此pH值范围内,羧基甜菜碱两性离子改性的色谱固定相的表面带正电荷[12]。羧基甜菜碱两性离子固定相在色谱领域最早报道是Elefterov等 [13]用赖氨酸类羧基甜菜碱两性离子固定相分离金属离子; Colman等[14]在硅胶基质C18整体柱表面键合羧基甜菜碱改性涂层,应用在离子色谱中,有效分离无机阴离子; Violeta等[15]使用交联羧基甜菜碱型两性离子固定相,选择性分离无机盐溶液中多种二,三价的重金属离子。而羧基甜菜碱型功能单体在亲水作用色谱领域中罕有报道。
表面引发原子转移自由基聚合(Surfaceinitiated atom transfer radical polymerization, SIATRP)技术是近年发展起来的一种新型聚合技术,可在基材表面形成高密度聚合物分子链[16],且兼具活性聚合和接枝链长可控等优点[17],在色谱固定相的制备方面也得到广泛应用。Pan等[18]通过SIATRP技术制备将2甲基丙烯酸3(2,4,5三羟基3氨基6羟甲基四氢吡喃)聚合物链接枝在硅胶微球表面,形成壳层结构,可提供更多结合位点,有效固定植物凝聚素且提高了负载量; Takuya等[19]通过SIATRP技术将N异丙基丙烯酰胺接枝在聚苯乙烯整体柱上,与接枝N异丙基丙烯酰胺的空心毛细管柱相比,在水溶液环境中更有效地分离地塞米松,可的松等生物活性溶质。
本研究使用甲基丙烯酰氧乙基二甲基乙酸铵(CBMA)作为功能单体,采用SIATRP技术,以硅胶为基质,在键合引发剂的硅胶表面进行聚合反应,通过改变功能单体的加入量,制备了接枝量不同的SilicaCBMA亲水作用色谱固定相。使用有机酸类化合物作为探针,研究了流动相pH值、盐浓度、水含量等因素对溶质在SilicaCBMA固定相上保留的影响,建立了芦丁片中维生素C含量的HILIC测定方法。
2 实验部分
2.1 仪器与试剂
岛津LC20AT(SPDM20A 二极管阵列检测器)高效液相色谱仪(日本岛津公司); PE Frontier 红外光谱仪(美国珀金埃尔默公司); Bruker Avance 400 MHz核磁共振仪(瑞士Bruker公司); KQ3200E型超声波清洗器(昆山市超声仪器有限公司); BS224S型电子天平(德国赛多利斯科学仪器有限公司); V77192US型高压气动泵(德国柏林诺尔公司); Vario EL Ⅲ型元素分析仪(德国Elementar 公司)硅胶(粒径:10 μm,平均孔径: 10 nm,苏州纳微科技有限公司); 氯乙酸钠、甲基丙烯酸二甲氨基乙酯(DMAEMA)、2,2′联二吡啶(Bpy)、溴化亚铜(CuBr)、三乙胺(TEA)、2溴异丁酰溴、苯甲酸、对甲苯磺酸、阿魏酸、肉桂酸、维生素C(分析纯,阿拉丁试剂(上海)有限公司); 乙腈(ACN,色谱级,美国Sigma公司); 甲酸铵(分析纯,广州化学试剂厂); 四氢呋喃(THF,加入金属钠回流6 h,干燥备用)、甲苯(加入金属钠回流6 h,干燥备用)、浓氨水、甲醇(分析纯,天津市大茂化学试厂); 复方芦丁片(国药准字:H31021092; 上海朝晖药业有限公司)。
2.2 实验方法
2.2.1 甲基丙烯酰氧乙基二甲基乙酸铵功能单体的制备 功能单体参照文献[20]制备。将5.505 g氯乙酸钠以适量的去离子水溶解,用NaOH调节至pH 7.0,置于恒压滴液漏斗中。将7.704 g DMAEMA以少量去離子水溶解后置于250 mL三口圆底烧瓶中,全程通N2保护,50℃时开始滴加氯乙酸钠溶液,约10 min滴完,升温到60℃反应3 h。反应完毕,向圆底烧瓶加入乙醇,振摇后抽滤,将滤饼溶解在甲醇中,重结晶,得到甲基丙烯酰氧乙基二甲基乙酸铵(CBMA)功能单体,共计4.429 g,产率67.5%,见图1。
2.2.2 用SIATRP技术制备SilicaCBMA固定相 称取0.5 g CBMA功能单体溶于70 mL甲醇中,置于100 mL圆底烧瓶。将3.0 g溴代硅胶(参照文献[21]制备)、 0.166 g CuBr、 0.362 g 2,2′联吡啶置于200 mL圆底烧瓶中,将两个烧瓶密封充氮气30 min后,溶解CBMA的甲醇溶液转移至含溴代硅胶圆底烧瓶中,在N2保护下, 60℃反应24 h,反应结束后抽滤,将滤饼依次用大量0.1 mol/L EDTA2Na溶液、甲醇、丙酮洗涤多次,60℃真空干燥过夜,即得SilicaCBMA固定相,反应步骤见图2。
2.2.3 色谱柱的填充 称取2.0 g上述合成的固定相,以甲醇为匀浆液和顶替液,超声分散均匀后,在40 MPa压力下装入不锈钢柱管(150 mm × 4.6 mm I.D.)中。
2.2.4 液相色譜条件 流动相为乙腈甲酸铵溶液(9∶1, V/V),甲酸铵溶液浓度10 mmol/L,pH=4.0,流动相流速1.0 mL/min; 柱温25℃; 进样体积20 μL; 检测波长为254 和355 nm。
3 结果与讨论
3.1 单体的表征
3.1.1 功能单体的核磁表征 1HNMR(D2O, 400 MHz) δ: 6.00 (d, H, a); 5.61 (d, H, b); 1.78 (t, 3H, c); 4.47 (t, 2H, d); 3.95 (t, 2H, e); 3.16 (s, 6H, f)。目标产物的结构式如下,其特征位移化学峰与文献[20]相符。
3.1.2 功能单体的红外表征 FTIR (KBr)如图3所示, 338203 cm1处为OH 的伸缩振动吸收峰; 1713.79 cm1处为酯羰基的伸缩振动峰; 1134.50 cm1处为酯 COC的伸缩振动峰; 原料 ClCH2COONa 在 769.96 cm1处的CCl 伸缩振动峰消失; 915.34 cm1处的吸收峰是羧基甜菜碱中R3N+的吸收峰,证明ClCH2COONa 与 DMAEMA 进行了季铵化反应。在1382.03和1610.07 cm1处出现了COO对称伸缩振动峰和COO不对称伸缩振动峰,与文献[20]相符。
3.2 不同接枝量的SilicaCBMA固定相制备及表征
合成3种不同CBMA单体接枝量的SilicaCBMA固定相, 研究接枝聚合物接枝量对固定相性能的影响。在SIATRP反应中,通过改变单体加入量、聚合时间、引发剂浓度等条件,可以控制聚合物链长。本实验选择保持引发剂浓度、聚合时间等其它条件不变,只改变单体的浓度,按照催化剂、配体与单体的物质的量比分别为1∶2∶1、1∶2∶2、1∶2∶4制备得到了CBMA单体接枝量不同的固定相SilicaCBMA 1、SilicaCBMA 2和SilicaCBMA 3。
式(1)[22]中, Cp是接枝聚合后增加的碳百分含量; Cp(calcd.)是单体分子中碳的理论百分含量; Ci 是固定引发剂后增加的碳含量; Ci(calcd.)是引发剂单元分子中碳的理论百分含量; A 是硅胶的比表面积。
利用元素分析对制备的3种SilicaCBMA固定相进行表征,由式(1)计算CBMA功能单体接枝量,结果见表1,接枝CBMA单体后的SilicaCBMA固定相与溴代硅胶相比,C, H和N 3种元素的含量明显增加,表明CBMA单体已经成功接枝在硅胶表面,且接枝聚合物的链长随初始单体浓度的增加而增长[23]。
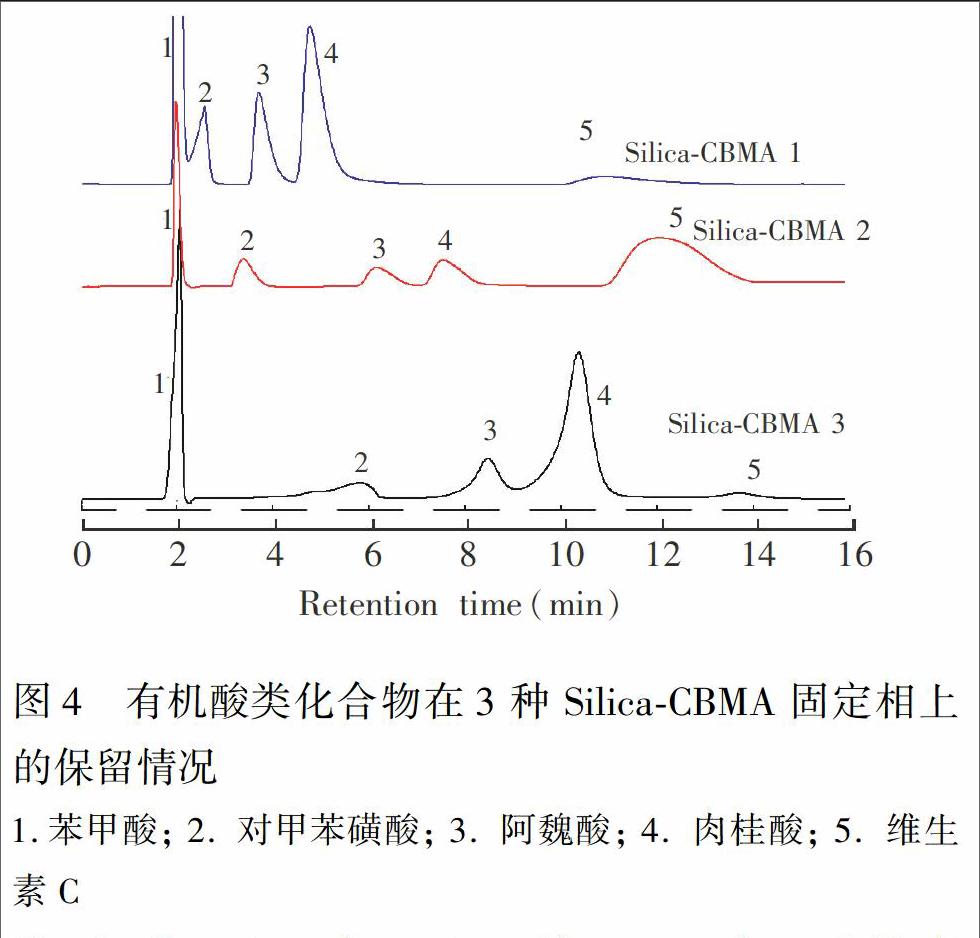
3.3 SilicaCBMA固定相接枝量对柱效的影响
以有机酸类化合物为探针化合物,在流动相pH=6的条件下,研究CBMA功能单体接枝量对分离性能的影响。随着CBMA单体的接枝量增大,溶质的保留时间延长。这是因为在实验条件下,SilicaCBMA固定相带正电荷[12],且表面正电荷密度随两性单体的接枝量增加而增大[24],酸性溶质电离带负电荷,所以溶质的保留时间随单体的接枝量增加而延长。但接枝量过大时,溶质与固定相作用力增加会导致峰形变差,结果见图4。
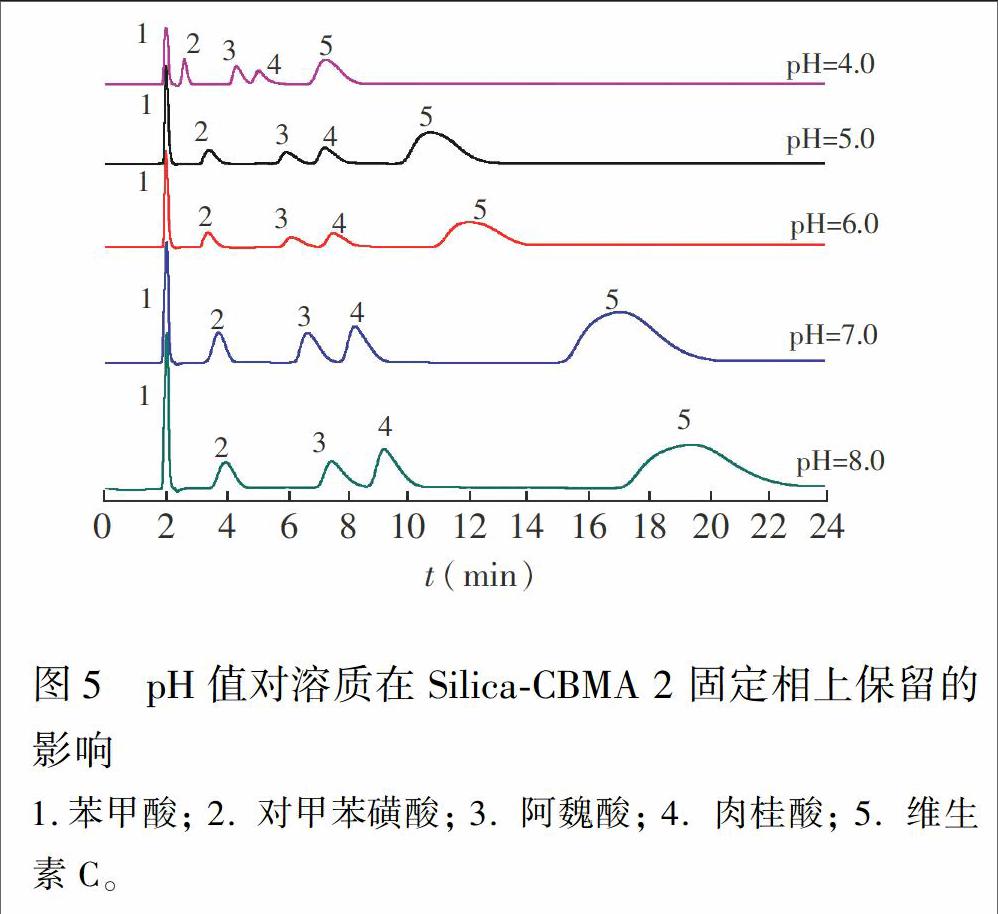
3.4 pH值对溶质保留的影响
在流动相pH值4.0~8.0的范围内考察有机酸类化合物的保留情况。由图5可见,酸性溶质保留时间都随着甲酸铵溶液pH值增大而延长,且在3种SilicaCBMA固定相上保留规律相同。这是因为在甲酸铵溶液pH值从4.0增至8.0的过程中,SilicaCBMA固定相功能基电离程度变大[25],酸性溶质电离且极性也变大[26],二者间的静电作用增强使得溶质保留增强。
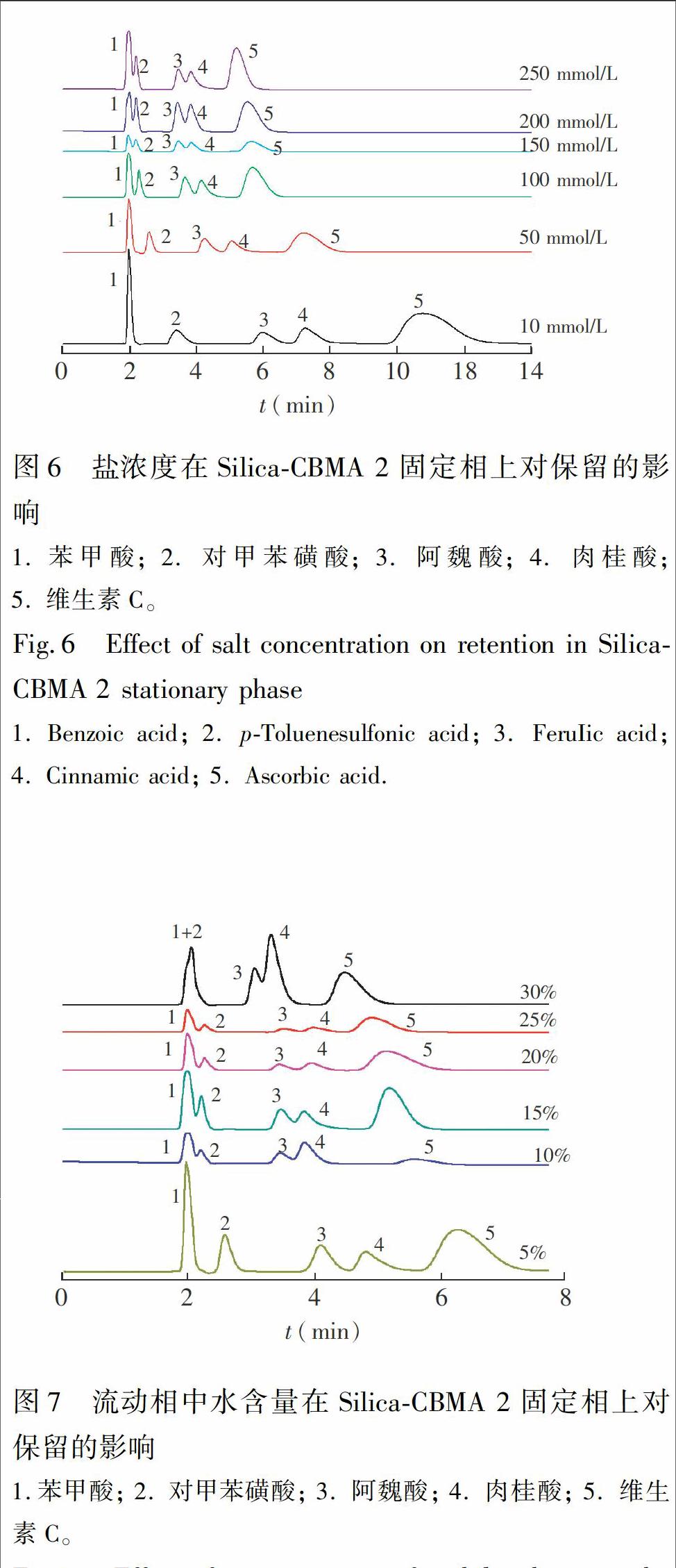
3.5 盐浓度对溶质保留的影响
在甲酸铵溶液浓度10~250 mmol/L的范围内考察了有机酸类化合物的分离情况。由图6可见,随着甲酸铵溶液浓度的提高,5种溶质保留时间都随之降低,且在3种不同接枝量的SilicaCBMA固定相上保留规律相同。这是因为,固定相表面带正电荷,酸性溶质电离带负电荷,两者之间存在离子交换作用,溶质保留随流动相盐浓度提高而减弱符合离子交换色谱的特征[27]。
3.6 水含量对溶质保留的影响
在流动相pH=5.0条件下改变流动相水含量(5%~30%),同时保持流动相中甲酸铵总浓度为10 mmol/L不变,探测水含量对有机酸类化合物保留的影响。
由图7可见,流动相中乙腈与甲酸铵溶液的体积比从95∶5到70∶30,随着流动相强洗脱剂水含量的增加,酸性溶质保留都减弱,这是典型的亲水作用色谱特征,可见酸性溶质在此SilicaCBMA柱子上的保留是离子交换作用与亲水作用共同作用的混合模式[28]。
3.7 复方芦丁片中维生素C、芦丁含量的测定
复方芦丁片主要含有芦丁和维生素C两种成分,用于高血压脑病、脑出血、视网膜出血等症状的辅助治疗[29]。本研究采用自合成的SilicaCBMA亲水色谱固定相测定复方芦丁片中维生素C、芦丁的含量。
3.7.1 線性范围的测定 称取适量维生素C、芦丁对照品,分别用50 %甲醇定容为10~250 mg/L的对照品系列溶液。取上述对照品系列溶液,分别按照2.2.4节进样并平行测定3次,以质量浓度(mg/L)为横坐标,峰面积为纵坐标绘制标准曲线,维生素C标准曲线方程y=4196.5x + 106,相关系数r=0.9991, 线性范围为10~200 mg/L; 芦丁标准曲线方程y=33096.0x-17120,相关系数r= 0.9994,线性范围为20~120 mg/L。结果表明,维生素C、芦丁对照品溶液的质量浓度与峰面积呈良好的线性关系。
3.7.2 重复性实验 维生素C、芦丁供试品溶液的制备:取复方芦丁片5片,研细,精密称取相当于1片的质量,置于100 mL容量瓶中,加适量50%甲醇超声溶解后,再用50%甲醇定容,用45 μm微孔滤膜过滤。分别准确移取5 mL滤液于5个50 mL容量瓶中,用50%甲醇定容,作为供试品溶液。
测得5份供试品溶液中维生素C含量为55.5 mg,RSD为1.5%; 芦丁的含量为40.3 mg,RSD为1.8%,表明本方法测定芦丁片中维生素C、芦丁含量的重复性良好。
3.7.3 加标回收实验 取3.7.2节的2份供试品溶液,分别加入浓度不同的3组维生素C、芦丁对照品溶液,每组平行测定3次,计算加标回收率,结果见表2。
4 结 论
本研究采用SIATRP技术, 将CBMA接枝到硅胶表面, 通过控制反应条件,得到了CBMA不同接枝量的SilicaCBMA固定相,此固定相可有效分离有机酸类化合物。对酸性溶质而言,溶质在固定相上的保留随键合量增大而增强。酸性溶质在固定相上的保留是亲水作用和离子交换作用共同起作用的混合模式。溶质的保留随流动相盐浓度增大而减弱,随流动相pH值增大而增强,符合离子交换色谱的特征; 溶质的保留随流动相中水含量提高而减弱,表现出亲水作用色谱的保留特征。将所得固定相应用于芦丁药品中维生素C、芦丁含量的测定,操作方法简单,线性关系、精密度良好,为强极性样品的分离提供了新方法。
References
1 Alpert A J. J. Chromatogr. A, 1990, 499(19): 177-196
2 YANG Duo, YU DongPing, DONG XueFang, SHEN AiJin, JIN GaoWa, GUO ZhiMou, YAN JingYu, LIU MingYang, LIANG XinMiao. Chinese J. Anal. Chem., 2015, 43(10): 1439-1444
杨 铎, 俞冬萍, 董雪芳, 沈爱金, 金高娃, 郭志谋, 闫竞宇, 刘名扬, 梁鑫淼. 分析化学, 2015, 43(10): 1439-1444
3 Getu K, Song H Y, Ann V S, Deirdre C, Erwin A. J. Pharmaceut. Biomed., 2014, 87(18): 142-154
4 Bekir C, Begum O, Cigdem K. Chromatographia, 2014, 77(21): 1511-1520
5 Yu D P, Guo Z M, Shen A J, Yan J Y, Dong X F, Jin G W, Long Z, Liang L N, Liang X M. Talanta, 2016, 161(1): 860-866
6 Knox J H, Jurand J. J. Chromatogr. A, 1981, 218(20): 341-354
7 Pohlentz G, Marx K, Mormann M. Methods Mol. Biol., 2015, 1394: 163-179
8 Montserrat M A, Estela G, Jose B, Victoria S N. Anal. Chim. Acta, 2016, 940: 92-103
9 Luan X, Gao Y G, Guan Y Y, Jonathan F L, Zhao M, Lu Q, Liu Y R, Liu H J, Dong X, Yang S C, Zheng L, Sun P, Fang C, Chen H Z. Biomaterials, 2016, 95: 60-73
10 Qiu D Y, Li F, Zhang M Y, Kang J W. Electrophoresis, 2016, 37(12): 1725-1732
11 Shao Q, Jiang S Y. Adv. Mater., 2015, 27(1): 15-26
12 Hu F L, Chen K M, Xu H, Gu H C. Colloid. Surface. B, 2015, 126: 251-256
13 Elefterov A I, Kolpachnikova M G, Nesterenko P N, Shpigun O A. J. Chromatogr. A, 1997, 769(2): 179-188
14 Colman O R, Leon B, Ekaterina N, Pavel N N, Brett P. J. Chromatogr. A, 2006, 1109: 111-119
15 Violeta N, Silvia V, Stefania R. Chem. Eng. J., 2010, 162: 965-973
16 MA MeiHua, WANG XiaoZhong, GONG YanRu, NIU YuLing, WANG Yue, WANG HuiJun, LUO RuiMing, GONG BoLin. Chinese J. Anal. Chem., 2015, 43(3): 379-386
馬梅花, 王晓中, 龚艳茹, 牛玉玲, 王 玥, 王惠军, 罗瑞明, 龚波林. 分析化学,2015, 43(3): 379-386
17 Wang H S, Song M, Hang T J. ACS Appl. Mater. Interfaces, 2016, 8: 2881-2898
18 Pan Y T, Bai H H, Ma C, Deng Y L, Qin W J, Qian X H. Talanta, 2013, 115(15): 842-848
19 Takuya K, Taka A A, Akihiko K. Colloid. Surface. B, 2016, 147: 408-415
20 Zhang L M, Tan Y B, Huang S J, Chen D Q, Li Z M. J. Macromol. Sci. A, 2000, 37(10): 1247-1260
21 Aya M, Kenichi N, Akihiko K, Hideko K, Yoshikatsu A, Jun K, Masahiko A, Teruo O. J. Chromatogr. A, 2010, 1217(4): 522-529
22 Farnoosh R, Markus A, Maria M T. J. Chromatogr. A, 2008, 1203(2): 160-167
23 Hao J X, Wang F Q, Dai X J, Gong B L, Wei Y M. Talanta, 2011, 85(1): 482-487
24 Lezova A V, Vlasovb P S, Lezova A A, Domninab N S, Polushinaa G E. Poly. Sci. A, 2011, 53(11): 1012-1018
25 Li Y, Liu R Y, Shi Y J, Zhang Z Z, Zhang X. Theranostics, 2015, 5(6): 583-596
26 Tang S, Wang L C, Han H F, Qiu H D, Liu X, Jiang S X. RSC Adv., 2013, 3: 7894-7901
27 Wang J X, Guo Z M, Shen A J, Yu L, Xiao Y S, Xue X Y, Zhang X L, Liang X M. Anal. Chim. Acta, 2015, 1398: 29-46
28 Tark A, Hayriye A, Berrin Z, Recep Z. Talanta, 2015, 131: 64-73
29 Li S M, Yang B B, Wang J, Bin D, Wang C Q, Zhang K, Du Y K. Anal. Methods, 2016, 8: 5435-5440
