LiFePO4电化学反应机理、制备及改性研究新进展
2017-06-21张英杰朱子翼邱振平梁慧新
张英杰 朱子翼 董 鹏 邱振平 梁慧新 李 雪
(昆明理工大学,锂离子电池及材料制备技术国家地方联合工程实验室,云南省先进电池材料重点实验室(筹),昆明 650000)
[Review]
LiFePO4电化学反应机理、制备及改性研究新进展
张英杰 朱子翼 董 鹏 邱振平 梁慧新 李 雪*
(昆明理工大学,锂离子电池及材料制备技术国家地方联合工程实验室,云南省先进电池材料重点实验室(筹),昆明 650000)
作为用于可持续能源的有效能量存储装置,锂离子电池因具有优异的电化学性能而得到广泛研究,是非常有发展潜力的储能电池体系,其技术发展及应用的关键在于电极材料的研发。LiFePO4作为锂离子电池正极材料之一,具有循环寿命长、能量密度大、充放电平稳、热稳定性良好、安全性好、重量轻和低毒性等优点,备受国内外专家的专注。然而,LiFePO4正极材料的研究还存在一些技术瓶颈,由于其存在电导率相对较低、锂离子扩散系数小以及振实密度不高等问题,导致循环性能、低温特性和高倍率充放电性能等并不理想,因而制约着它的应用和发展。近几年研究工作者通过改进制备工艺以及进行相关改性研究,旨在逐步解决上述问题。本文简要综述了LiFePO4正极材料的最新研究成果,就其结构特征、电化学反应机理、制备方法和改性进行了系统介绍。探讨了目前LiFePO4正极材料面临的主要问题及可能的解决策略,并对其未来的研究方向和应用前景进行了展望。
LiFePO4;研究进展;电化学反应机理;制备方法;改性
Key Words: LiFePO4; Research progress; Electrochemical reaction mechanism; Preparation method; Modification
1 引 言
自1997年Goodenough等1发现了具有橄榄石型结构的LiFePO4,经过专家、学者数十年富有成效的研究,其制备和改性工艺日趋成熟。与传统的锂离子电池正极材料相比,LiFePO4因其具有稳定的结构、较高的工作电压(3.45 V (vs Li/Li+))、理论放电比容量高(~170 mAh·g-1)、循环寿命长且原料资源丰富、价格便宜、绿色环保无污染等特性,是最有希望应用于航空航天、电动汽车(EVs)和混合动力电动汽车(HEVs)等重要领域的新一代锂离子电池正极材料之一2-7。
作为聚阴离子正极材料,LiFePO4存在电导率(10-9-10-10S·cm-1)和锂离子扩散速率(10-14-10-16cm2·s-1)偏低的缺点,导致最初合成的纯LiFePO4在0.05 mA·cm-2电流密度下的放电比容量仅为100-110 mAh·g-1,与理论数值相差较大。研究工作者还发现,纯LiFePO4在1C倍率下循环15次后的容量衰减高达20%以上,在高倍率下的放电比容量也极低,且-20 °C低温下0.5C倍率的放电比容量只有室温下的50%,其循环性能、低温特性和高倍率充放电性能等有待优化1,8-10。

张英杰,1963年生。1999年博士毕业于昆明理工大学有色金属冶金专业。现为昆明理工大学冶能学院博士研究生导师、教授。主要研究方向为电化学防护与环保、电化学能源。主持省部级科研项目17项。
董鹏,1980年生。2011年博士毕业于昆明理工大学冶金物理化学专业。现为昆明理工大学冶能学院讲师。主要研究方向为金属防腐与防护。

梁慧新,1992年生。2014年本科毕业于江苏大学材料学院材料成型专业,2014年至今为昆明理工大学冶能学院冶金工程专业硕士研究生。主要研究方向为锂离子电池正极材料。

朱子翼,1991年生。2013年本科毕业于福建工程学院材料学院材料成型专业,2015年至今为昆明理工大学材料学院材料工程专业硕士研究生。主要研究方向为锂离子电池正极材料。

邱振平,1990年生。2013年本科毕业于兰州理工大学材料学院焊接技术与工程专业,2013年昆明理工大学材料学院材料物理与化学专业。主要研究锂离子电池NCA正极材料的制备与改性。

李雪,1985年生。2015年博士毕业于厦门大学物理化学专业。现为昆明理工大学冶能学院讲师。主要研究方向为先进二次电池及相关能源材料,包括锂离子电池和钠离子电池。主持国家自然科学基金1项。
近年来,随着各种改善LiFePO4正极材料性能研究的深入,其实际充放电比容量已接近理论值,且循环稳定性、低温特性和高倍率性能都得到了较大的提升,LiFePO4正极材料产业化进程也明显加速。LiFePO4正极材料产业发展的历史不长,总体来说还处于初始阶段。目前,新能源汽车、储能设备、电动工具及电动自行车是LiFePO4电池最主要的四个应用领域。根据LiFePO4正极材料的用途不同,可分成两种不同的类型:容量型和功率型。容量型侧重高比容量,追求较高振实密度和压实密度,适用于低电流放电的电池(小型动力电池),一般倍率要求约3C以下;功率型侧重高功率,大电流放电电池(大型动力电池、储能电池),倍率要求5C-10C或以上。从供给方面来看,国外生产LiFePO4正极材料性能较好的企业有美国Valence、加拿大Phostech等,但他们的制备条件苛刻,生产成本较高。国内在LiFePO4正极材料领域的产业化起步并不晚,现阶段的生产企业已有近百家,诸如天津斯特兰、北大先行、博骏科技、云南汇龙等,不过多数尚处在研发阶段(包括小试和中试),能够实现批量生产的企业不多,且绝大部分还处在小批量试产阶段。值得一提的是,万向A123公司已经开发出了超级纳米LiFePO4技术,可以提高电池功率的50%-60%。说明产业化LiFePO4正极材料性能的改善空间很大,正是大力切入的大好时机。此外,中国政府对LiFePO4电池产业的发展给予了极大的关注,科技部于2009年在863计划中单独设立“磷酸铁锂正极材料规模化生产和应用关键技术研究”重点项目,支持资金上千万元,2008-2009年工业与信息化部先后设立“磷酸铁锂正极材料研发及产业化”与“磷酸铁锂动力电池及系统集成技术研发及产业化”重点专项,面向全国招标,对LiFePO4正极材料的研发及产业化给予重点扶持。本文从结构特征、电化学反应机理、制备方法和改性等方面,系统综述了近年来LiFePO4正极材料研究的最新进展。

图1 LiFePO4的晶格结构12Fig.1 Lattice structure of LiFePO412Adapted from Elsevier Science Publisher.
2 LiFePO4的结构特征
LiFePO4为橄榄石型结构,属于正交晶系,空间群为Pnmb。每个晶胞中有4个LiFePO4单元,其晶胞参数为:a = 0.6010 nm,b = 1.0332 nm,c = 0.4692 nm11。图1为LiFePO4的晶体结构示意图12。在LiFePO4中,氧原子以轻微形变的六方紧密堆积方式排列,磷原子占据氧原子四面体4c位,形成PO4四面体。铁原子和锂原子分别占据氧原子八面体的4c位和4a位形成FeO6八面体和LiO6八面体。交替排列的FeO6八面体、LiO6八面体和PO4四面体形成层状脚手架结构。在bc面上,相邻的FeO6八面体通过共用顶点的一个氧原子相连,形成FeO6层。在FeO6层之间,相邻的LiO6八面体在b方向上以共用棱上的两个氧原子相连成链。而每个PO4四面体既与一个FeO6八面体共用棱上的两个氧原子,又与两个LiO6八面体共用棱上的氧原子。Li+在4a位形成共棱的连续直线链,并平行于c轴,使Li+具有二维可移动性,在充放电过程中可以脱出和嵌入。而强的P―O共价键形成离域的三维立体化学键,使LiFePO4具有很强的热力学和动力学稳定性。但是,在LiFePO4结构中由于没有连续的FeO6共边八面体网络,不能够形成电子导体,电子传导只能通过Fe―O―Fe进行,故LiFePO4电子导电率低。同时,由于FeO6八面体和LiO6八面体之间的PO4四面体限制了晶格体积的变化,导致Li+的脱嵌运动受到影响,造成LiFePO4的锂离子扩散速率较低13-17。
3 LiFePO4的电化学反应机理
LiFePO4充放电过程中电极反应如下所示1,18,19。
充电反应:
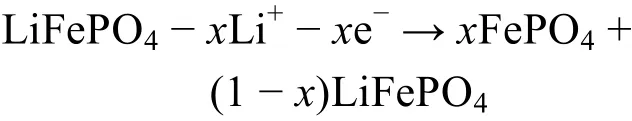
放电反应:

总反应:
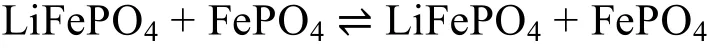
在LiFePO4正极材料中Li+的脱/嵌反应发生在LiFePO4/FePO4两相界面,使得正极材料的充放电曲线平整且电压电位稳定。理想的锂离子电池反应均一性良好,无任何副反应发生。实际上锂离子电池的充放电过程要复杂的多,导致其性能达不到预期效果,只有将这些复杂反应的机理研究清楚,才能进一步提高锂离子电池的性能。目前关于LiFePO4的电化学反应机理认识主要有以下三种观点。
3.1 单纯两相型反应机理
不同于其他正极材料倾斜的充放电曲线,LiFePO4正极材料的充放电曲线在3.45 V (vs Li/Li+)的电位平台相对平整,因此Padhi等1认为LiFePO4的脱/嵌过程是Li+在LiFePO4/FePO4两相界面的脱出/嵌入过程,即两相反应。
3.1.1 收缩核模型
为了描述这种两相反应的微观机制,解释LiFePO4的充放电机理,Srinivasan等20认为在两相共存区域之外有一个相应的单相区域,并建立了一个LiFePO4锂离子电池中锂插层和相变的数学模型,由此提出了简单的收缩核模型,用于描述Li+嵌入脱出过程中两相共存界面的变化。图2为具有两相并置和相边界运动的收缩核模型示意图,该模型解释了不同倍率下LiFePO4充放电过程中的电压/容量曲线。遗憾的是,这个简单收缩核模型并未考虑到由离子运动受橄榄石型结构约束所引起的任何各向异性。
3.1.2 新核-壳模型
Laffont等21通过电子能量损失谱观测平板状LiFePO4颗粒的微观形貌,分析认为纳米尺度的界面区域是由LiFePO4和FePO4两相组成,而不是固溶体。在脱锂过程中,FePO4的形核区在颗粒内部沿a轴方向延伸至整个晶粒,反之在嵌锂过程也同样适用。图3为新核-壳模型示意图。但是,由于他们的高分辨电子能量损失谱测量并未在原位完成,没有得出是否存在暂态固溶体的明确结论。
3.1.3 多米诺模型
Delmas等22采用X射线衍射和电子显微镜技术研究了由传统固相法制备出的LiFePO4纳米颗粒的锂离子脱/嵌过程。发现部分的LiFePO4二次颗粒是由晶相Li1-yFePO4或者LixFePO4的单晶颗粒组成的,这表明在其它晶相的颗粒中,另一相的单晶生长速度要显著快于形棱速度的。他们建立了一个多米诺模型,如图4所示,用于解释在材料离子电导和电子电导率都很低的情况下,纳米级别结晶完好的颗粒可以进行大电流充放电的原因。
所以迄今为止还没有哪一种模型能够完美解释LiFePO4的充放电机理,但对其研究的热潮依然持续着。最近,Liu等23通过原位X射线衍射技术捕获到LiFePO4纳米颗粒电极高倍率循环过程中的亚稳态结构,如图5所示。这种非平衡易位相转变反应机理的研究为基于两相反应机理的锂离子电池正极材料的高倍率性能提供了新的研究思路。
图2 具有两相并置和相边界运动的收缩核模型示意图20Fig.2 Illustration of the shrinking-core model with the juxtaposition of the two phases and the movement of the phase boundary20
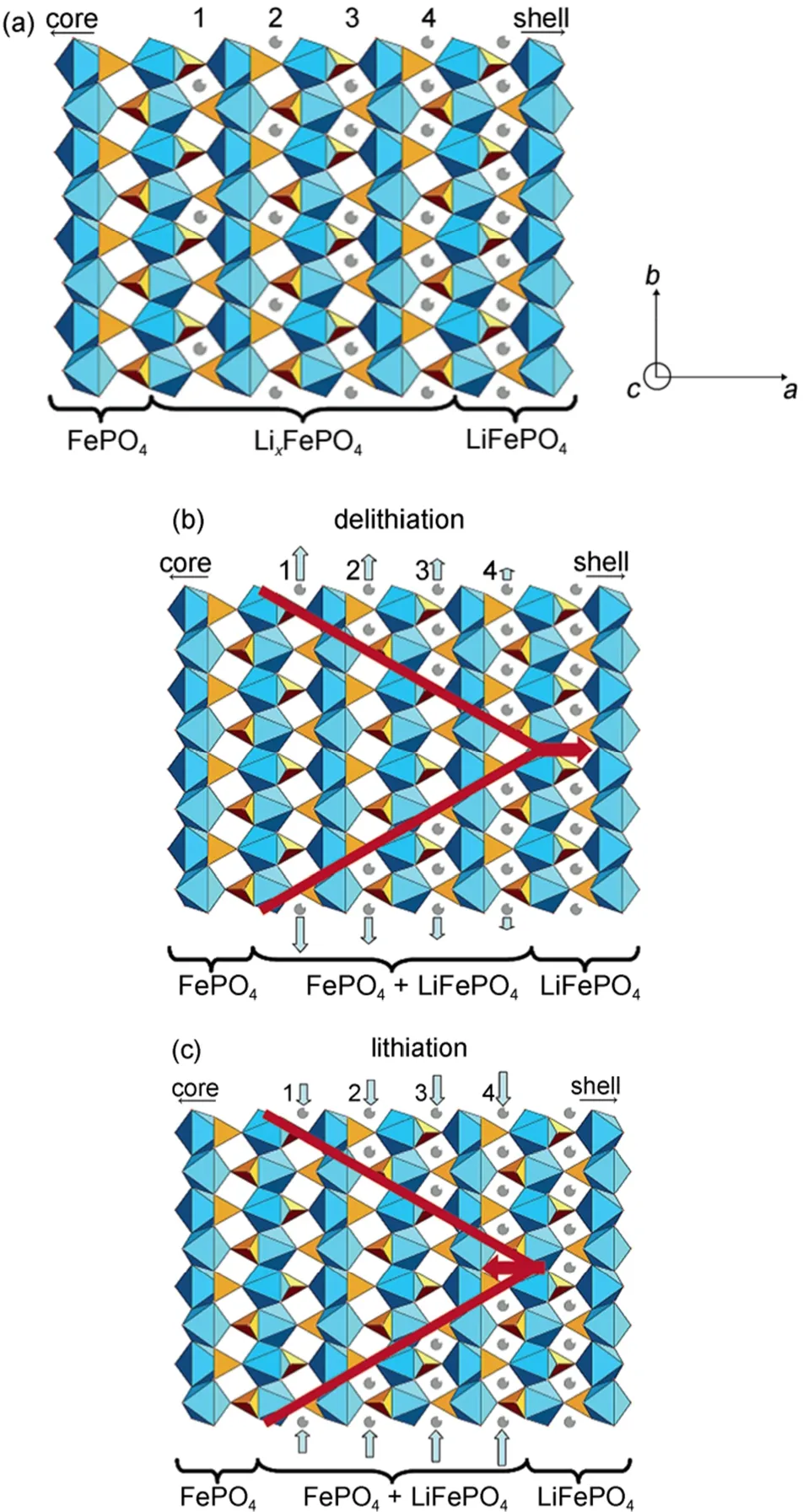
图3 LiFePO4和FePO4相之间的界面区域示意图21Fig.3 Schematic views of the interfacial region between LiFePO4and FePO4phases21
3.2 单相反应机理
不同于两相反应机理模型,有观点认为LiFePO4脱/嵌锂是单相反应。Gu等24采用静电纺丝法制备LiFePO4正极材料,通过球差校正环形明场扫描透射电子显微成像技术直接观察到LiFePO4正极材料中的锂离子,并在部分脱锂的LiFePO4单晶纳米线(d = 65 nm)观测到了锂离子隔行脱出现象,如图6所示,其中黄色圈为锂离子存在的位置,橘色圈为锂离子脱出的位置。这一结构与石墨插层化合物中出现的“staging-II,二阶”现象类似,与之前提出的各类两相反应模型均不一致,为单相结构。
Liu等25通过软X射线吸收光谱、硬X射线拉曼散射和理论模拟,深入、系统的研究了LiFePO4纳米颗粒和单晶中的电子结构的相变和锂化效应。同时,他们进行了第一性原理计算和光谱的多重模拟,定量确定通过完全锂化过程重新分配的三维价态。通过在LiFePO4单晶上收集的沿着锂扩散方向的偏振相关光谱,进一步验证了电子重构现象的发生。研究中发现,三维价态的演变总体上与局部晶格畸变一致,从而可以通过观察三维价态来确定LiFePO4在锂化过程中电子结构的变化过程,如图7所示。通过高质量光谱的两相拟合,他们检测到两相行为的微妙偏差,这一小偏差表示反应过程中存在有限量的非平衡中间相而不是固溶体相。
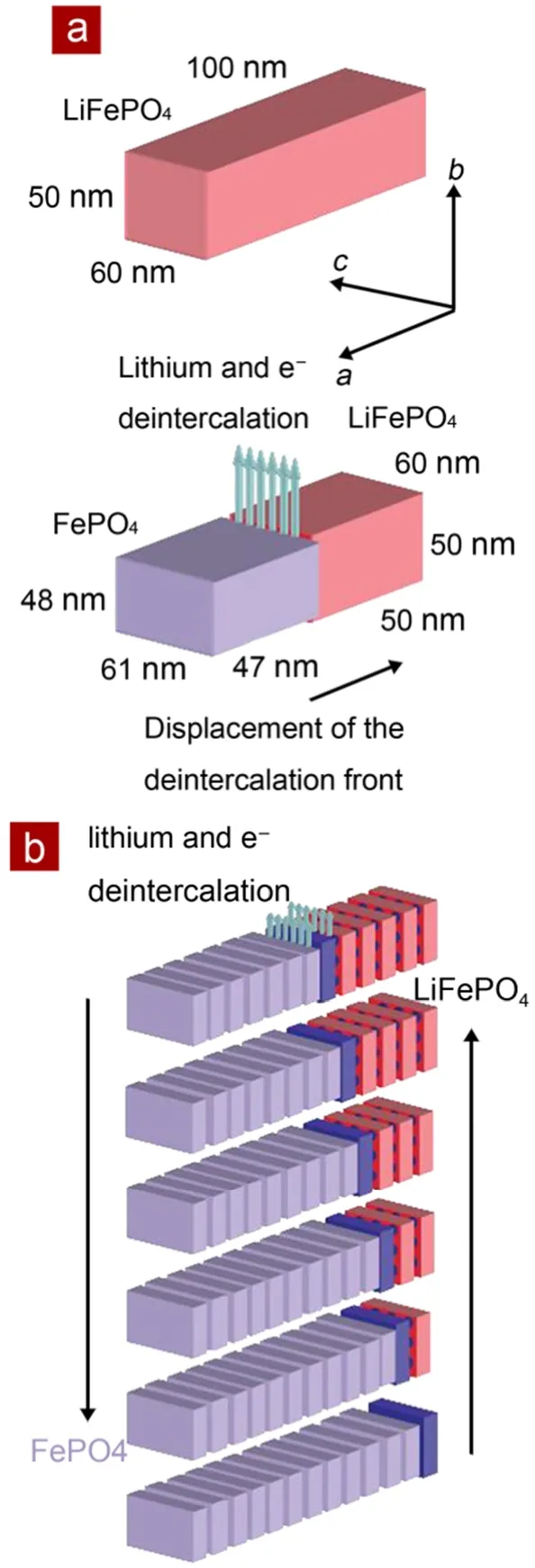
图4 LiFePO4晶体中锂脱嵌机理的多米诺模型示意图22Fig.4 Schematic view of the ‘domino-cascade’mechanism for the lithium deintercalation/ intercalation mechanism in a LiFePO4crystallite22
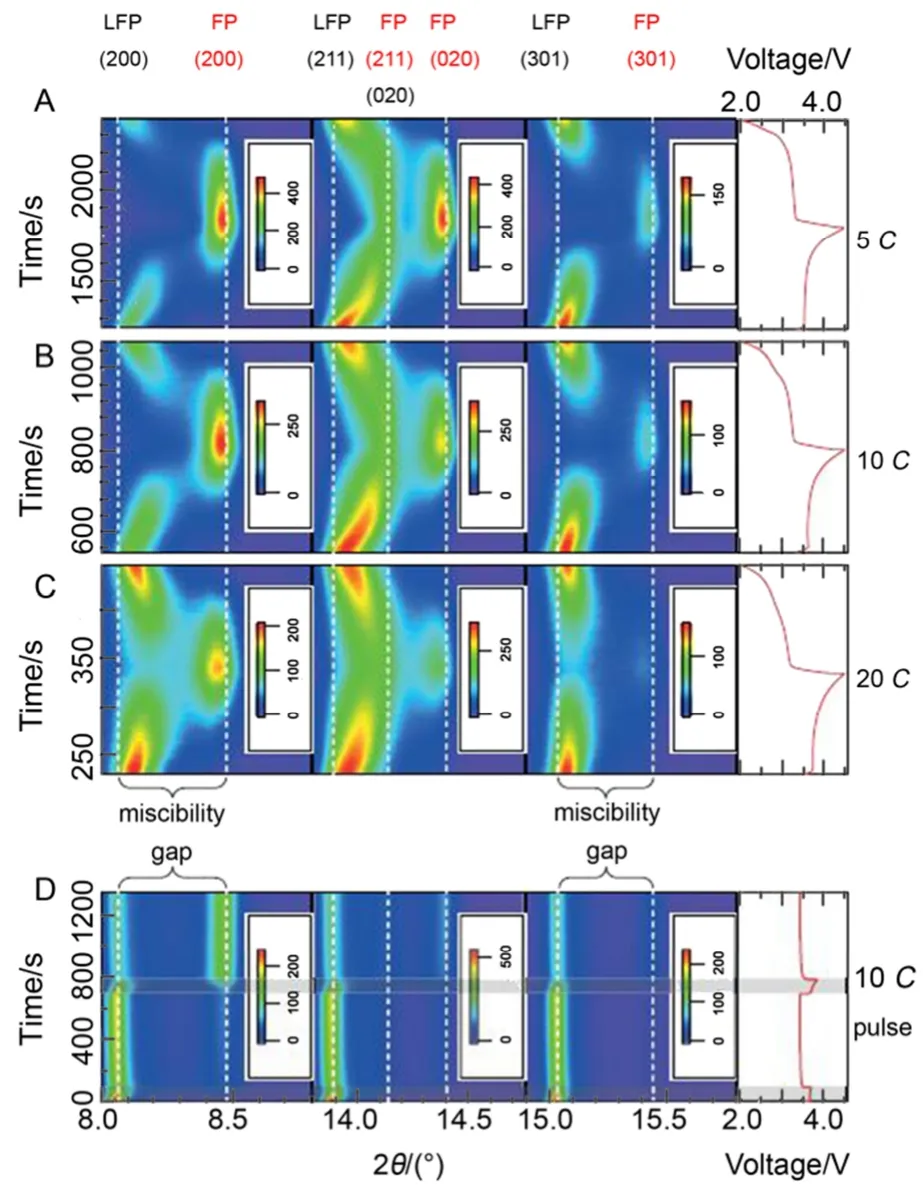
图5 LiFePO4在不同电化学循环条件下的原位XRD图23Fig.5 In situ XRD pattern of LiFePO4under different electrochemical cycling conditions23
Orikasa等26采用时间分辨的XRD对锂化过程中LiFePO4和FePO4之间的非平衡相变进行了实时跟踪,如图8所示。研究表明,随着电流密度的增加,除了LiFePO4和FePO4等热力学稳定的相之外,还会出现亚稳晶相。在开路状态下随着电化学循环的进行,亚稳晶相将逐渐减少,得出了LiFePO4脱锂过程中存在单相结构的结论。
3.3 三相反应机理
第三种观点认为LiFePO4在脱/嵌锂过程中出现LiFePO4/staging-II/FePO4三相共存结构。Sun等27采用密度泛函理论计算,证明了脱锂的LiFePO4中二阶结构是由锂离子传输动力学导致的热力学亚稳态结构,不同于石墨插层化合物中出现的“staging-II,二阶”现象。基于计算结果,他们提出了一种双界面模型来描述了LiFePO4在充放电过程中的脱/嵌锂机理,如图9所示。除了静电的直接相互作用,Fe2+/Fe3+的氧化还原对的间接相互作用使得锂离子只能采取隔行脱出途径,因为这样形成的“二阶”结构才在动力学上处于优势地位,然而热力学能量最低原理却支持两相分离反应机理。动力学与热力学条件的相互竞争导致LiFePO4颗粒在脱锂过程中出现LiFePO4/staging-II/FePO4三相共存结构,可以较好地解释实验现象28。但是,他们的研究没有考虑到尺寸效应的影响,而小尺寸颗粒的高比表面积可能改变反应条件,特别是热力学条件,这些都有待于进一步的研究证实。

图6 LiFePO4隔行脱锂的ABF像24Fig.6 ABF micrographs showing Li ions of partially delithiated LiFePO4at every other row24
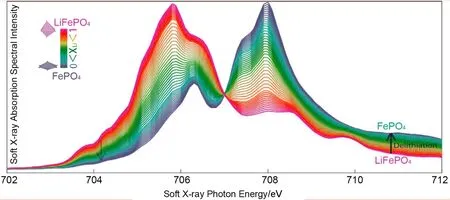
图7 软X射线吸收光谱图25Fig.7 Soft X-ray absorption spectra25
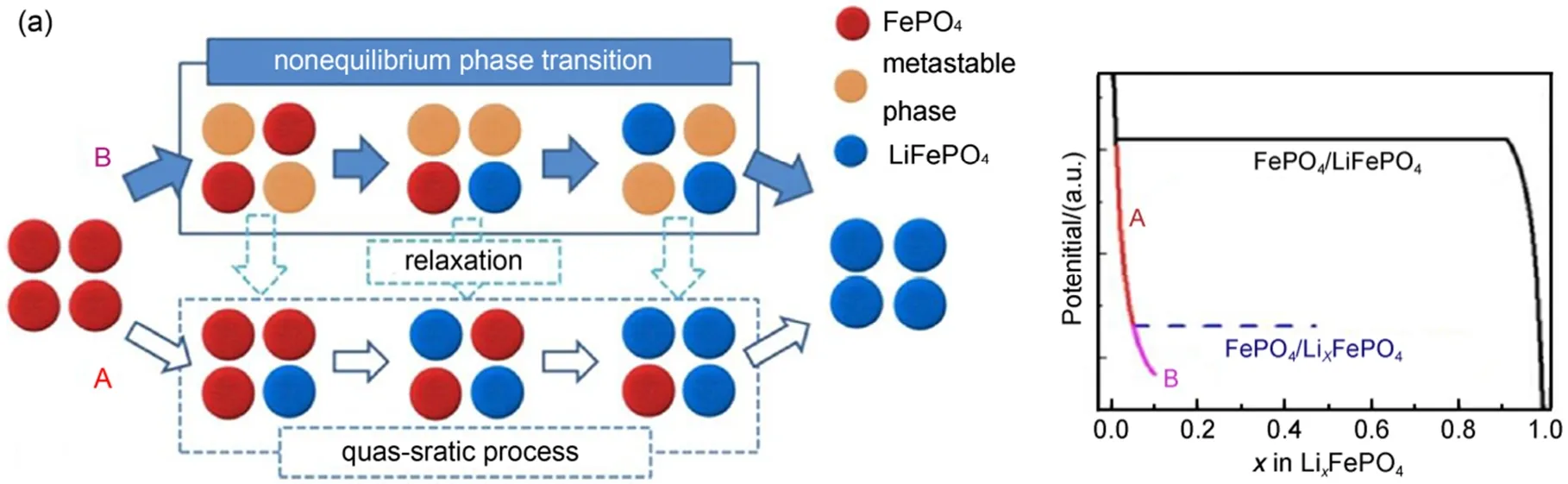
图8 时间分辨XRD测试和LixFePO4的电位变化曲线示意图26Fig.8 In situ time-resolved XRD test and schematic potential profiles for LixFePO426
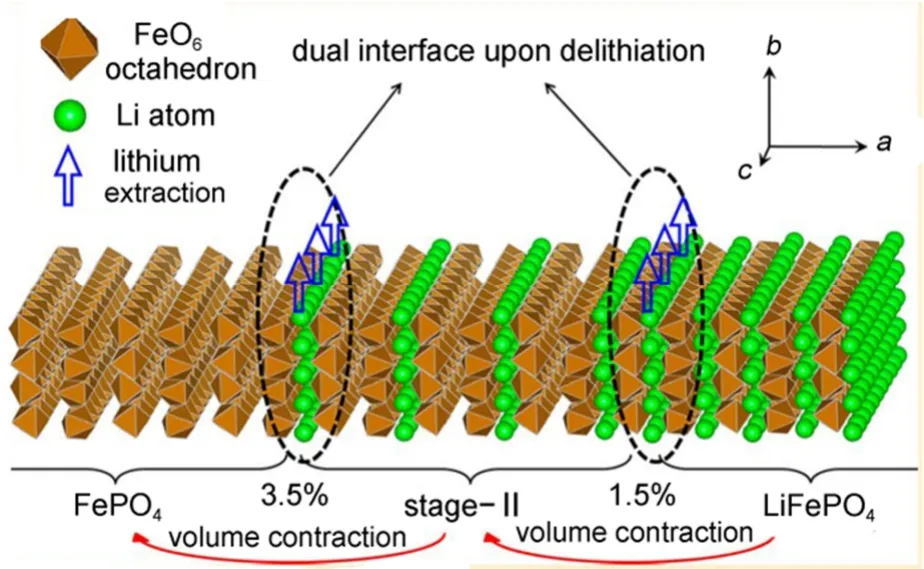
图9 具有二阶结构LiFePO4的脱锂双界面结构模型27Fig.9 Dual interface structure model with staging-II interface for delithiation of LiFePO427
4 LiFePO4制备的研究进展
自然界中的LiFePO4是以矿石的形式存在,但这些矿石中含有较多杂质,提纯工艺复杂,故需要通过人工合成的方式得到可以直接用作锂离子电池正极材料的LiFePO4。近年来,国内外专家学者致力于优化LiFePO4正极材料的制备工艺,以提升其性能,实现商业化生产的扩大应用。
4.1 传统的制备方法
高温固相法、碳热还原法、溶胶-凝胶法、水热法等作为最早应用于制备LiFePO4正极材料的方法,因其操作简单、成本较低、产物性能较好等优点,一直以来备受青睐,并作为传统的制备方法应用于产业化生产。但是,国内目前尚未出现真正的领军企业,只有少部分公司掌握着较为成熟的量产技术,行业缺乏原始创新技术,导致LiFePO4正极材料行业处于产业化临界点之下。因此,在现有的制备工艺基础上,提高原料纯度、优化原料配比和工艺参数等因素,提高LiFePO4正极材料的性能,使之扩大化生产显得尤为重要。
4.1.1 高温固相法
高温固相法是将原料按照一定化学比例进行研磨,混合均匀后在通有保护气氛下进行高温煅烧,从而得到LiFePO4的一种制备方法。高温固相法的优点主要是工艺简单、产量较高、污染较低、易于产业化生产,但能耗较大、需控制亚铁氧化度、正极材料电化学性能不易控制。天津斯特兰、北大先行、河南华鑫等大多数国内公司都是采用这种方法。
近年来,研究人员通过不断改进高温固相法制备LiFePO4的工艺参数,合成出的LiFePO4正极材料具有较好的电化学性能。Cui等29以黄磷工业生产的副产品Fe-P合金为原料,将混合物在CO2气氛下高温煅烧,合成LiFePO4/C复合材料。在电化学性能测试中,以0.1C的倍率充放电,复合材料的初始放电比容量达到150.6 mAh·g-1,15次循环后,放电比容量仍保持146.6 mAh·g-1,容量保持率约为97.3%,说明产物具有较高的充放电比容量和良好的电化学反应可逆性。这种方法不仅简化了生产过程,进一步降低了制造成本,而且减少温室气体排放,对工业应用具有重要意义。Churikov等30采用高温固相法制备LiFePO4/C复合材料,通过在不同温度范围内进行实验数据与热力学计算的比较,研究了原料和碳源的热解机理。实验结果表明,草酸亚铁热解的中间产物包含有Fe3O4、Fe2O3和FeO,随着工艺参数的轻微变化,草酸盐分解的多种路径可以同时进行。同时,Li3PO4的形成仅在室温下研磨的共混物中检测到,Li3PO4和NH4PO3是在温度高达800 °C下合成锂铁矿的基础。研究还发现,选用柠檬酸作为有机前驱体,会在室温下形成新的单取代无水C6H7O7Li相,并估计了柠檬酸能够形成具有最大导电率的碳涂层的热处理条件。本课题组目前以自制草酸亚铁为原料,采用高温固相法制备LiFePO4/C复合材料,分析不同煅烧温度下所得正极材料的物相结构。实验结果表明,煅烧温度过低导致产物中含有少量没有反应完全的原料和副反应产物,而过高的煅烧温度使得LiFePO4晶粒粒径增大,综合加工性能不好。因而选用合适的煅烧温度是影响LiFePO4/C复合材料性能的关键因素。
4.1.2 碳热还原法
碳热还原法是以三价铁化合物为铁源,高温下碳源将Fe3+还原为Fe2+,从而实现LiFePO4制备的一种方法。这种方法操作简单,使用较为廉价的三价铁源以节约成本,碳源既起到还原的作用,又在LiFePO4产物表面形成原位包覆,从而提高正极材料的导电性能31。碳热还原法的主要优点表现为产物堆积密度高、批次稳定性好、正极材料电化学性能较好,但产物的性能过于依赖原料的品质。目前,越来越多的公司开始采用碳热还原法批量生产LiFePO4/C正极材料,如苏州恒正、深圳比亚迪、台湾立凯电能和美国Valence等。
最近,Xiao课题组等32以Fe2O3、NH4H2PO4和Li2CO3为原料,葡萄糖作为碳源,制备结构完好的LiFePO4/C复合材料。电化学性能测试中,在0.1C、1C和2C下分别具有153.8、128.3和121.0 mAh·g-1的倍率性能。应用电化学阻抗谱研究LixFePO4/C (0 < x < 1)电极的温度变化时发现,随着温度的升高,LixFePO4/C电极的交换电流密度增加、电荷转移电阻减小和锂离子扩散速率提升。同时,复合材料中锂离子的浓度影响着锂离子扩散速率,前者的减小导致后者的增加。此外,还研究了不同碳源对使用Fe2O3制备LiFePO4/C复合材料性能的影响33。实验结果表明,选用均苯四甲酸酐为碳源可以最有效地提高复合材料的电化学性能。在采用中等扫描速率循环伏安法(CV)研究LiFePO4/C电极时34,通过曲线拟合获得以不同速率扫描LiFePO4/C电极的CV氧化还原峰电势的极限值,以扫描速率和温度变化研究LiFePO4/C电极的可逆性。研究发现,在较高测试温度下LiFePO4/C电极将展现更好的电化学可逆性。Weng等35以自制β-FeOOH纳米棒为铁源,以葡萄糖为还原剂和碳源,采用碳热还原法制备LiFePO4/C复合材料。研究了Fe(III)原料的形态和碳的含量对产物的电化学性能的影响。实验结果表明,均匀的β-FeOOH纳米棒和适量的葡萄糖可以有效地控制颗粒的尺寸和分布。其中,以2.79% (w,质量分数)的碳含量合成的LiFePO4/C复合材料在0.1C、1C、10C和15C倍率下的初始放电比容量分别为158.8、144.3、111.0和92.9 mAh·g-1,显示出优异的倍率性能。Hu等36以蔗糖和乳化剂T-80为碳源,采用两步添加法制备LiFePO4/C复合材料。实验结果显示,产物中无Fe3+的出现,且在LiFePO4颗粒的表面上可获得厚度约为3.5 nm的均匀碳包覆层。同时,复合材料表现出优异的电化学性能,在0.1C倍率下的初始放电比容量达到159.4 mAh·g-1,循环50次后容量保持率为98%,具有良好的循环稳定性。
4.1.3 溶胶-凝胶法
溶胶-凝胶法是将无机物或金属醇盐等经过溶液、溶胶、凝胶进而固化,然后将凝胶经低温干燥和高温煅烧的一种方法。溶胶-凝胶法合成的LiFePO4正极材料不仅颗粒较小,而且具有良好的分散性,反应过程容易控制,合成温度较低,对设备的要求不高。但因干燥收缩大、合成周期长、对合成过程中的气氛有特殊的要求等,产业化生产难度大,鲜有公司作商业化生产,常用作实验室研究。
Dhindsa等37以CH3COOLi·2H2O、FeCl3和P2O5为原料,月桂酸为表面活性剂,采用溶胶-凝胶法制备LiFePO4正极材料。除了主要的LiFePO4相外,XRD、磁性测试和穆斯堡尔谱学还显示出样品存在不同量的原位Fe2P杂质相,Fe2P杂质相的量很大程度上取决于煅烧温度。研究表明,为了提高LiFePO4/C复合材料的电化学性能,控制合成过程中的还原环境和温度显得尤为重要。Reklaitis等38将FeC2O4·2H2O、Li2CO3溶解于HNO3中,加热并滴加柠檬酸溶液,再加入NH4H2PO4混合加热搅拌,产生的凝胶干燥后进一步高温煅烧,得到纯相LiFePO4。XRD和MS清晰地显示了具有正交对称性LiFePO4的形成。同时,XRD和MS也显示了三价铁离子的存在,他们归因于LiFePO4纳米结构位于外层的缺陷。Ziolkowska等39采用溶胶-凝胶法制备LiFePO4正极材料,通过将原位高温XRD和原位高温TEM数据与正极材料的电化学性能进行比较,研究了煅烧温度对LiFePO4正极材料的结构和电化学性能的影响。实验结果表明,随着煅烧温度的升高,原位XRD显示出LiFePO4正极材料的衍射峰变得更尖锐,粒径尺寸增大。原位高温TEM中LiFePO4颗粒表面和形态略微改变,拥有更多的孔洞结构。同时,在低温煅烧时,LiFePO4正极材料的不完全结晶导致其电化学活性较低。随着煅烧温度的升高,正极材料中杂相α-Fe2O3浓度降低,结晶度提高,电化学性能得以提升。但在900 °C下煅烧的LiFePO4正极材料表现出比容量的严重降低(~30 mAh·g-1),主要是由于过高的煅烧温度导致活性颗粒的附聚和粒径尺寸的过度增大,正极材料的离子导电性降低和电化学电池的电阻率增大。结果显示在800 °C下煅烧合成的LiFePO4正极材料获得的性能最为优异。
4.1.4 水热法
水热法是在密闭反应容器(如高压釜)中,以水溶液或蒸汽等流体作为反应体系,通过对反应体系加热、加压,创造一个相对高温、高压的反应环境,使通常难溶或不溶的物质溶解并且重结晶,再进行分离和热处理得到产物的一种方法,属液相化学的范畴。水热法不仅合成工艺简单,而且水热反应过程中不需要通保护气体,制得的LiFePO4正极材料形貌规则、晶粒粒径较小、分散性较好,但成本较高、高温高压下操作的危险系数较大、产物的堆积密度和压实密度较小。目前采用这种方法作生产的公司有宁波英特维、加拿大Phostech和韩国韩华等。
Xu等40基于阴离子配位多面体的生长单元模型和分子模拟,研究了LiFePO4正极材料在水热条件下的形成机理和生长规律。系统计算了生长单元的形成和脱水反应的结合能,以揭示晶体的形核过程。在晶体生长过程中,用阴离子配位多面体的生长单元在不同表面上的结合能解释了晶体的生长规律。较大的结合能意味着沿着该方向的生长速率更快,相应的晶面逐渐减小,甚至消失。如图10所示,不同晶面的有利生长单元的结合能的大小不同,使得生长速率呈V{011}> V{210}>V{002}> V{101}> V{200}≈ V{020}的顺序,符合水热条件下主要显示{020}、{200}和{101}面的生长规律。此外,研究表明水热条件下前驱体溶液的pH值对LiFePO4颗粒的阴离子配位多面体的生长单元的形成、成核、形态和晶体习性起重要作用。不同pH值的前驱体溶液获得的络合物也不同,导致LiFePO4颗粒晶面的暴露程度不同,进而影响晶面的有利生长单元的形成速率和稳定性,产物的颗粒形貌、晶体取向和电化学性能等表现出较大的差异。此外,Zhao等41以不同配料温度下合成的LiFePO4正极材料进行对比,研究了配料温度对水热法制备LiFePO4正极材料的影响。实验结果表明,通过控制配料温度可以制得高纯Li3PO4中间体并抑制前驱体中Fe(OH)3的形成,有利于获得结晶良好的LiFePO4正极材料。在最优化的30 °C下配料时,合成的LiFePO4正极材料以0.1C倍率充放电,初始放电比容量达到156 mAh·g-1,0.5C倍率下放电比容量为151 mAh·g-1,在10C的高倍率时比容量仍然有127 mAh·g-1,20次循环后容量保持率超过99%,显示了优异的电化学性能和循环稳定性。水热反应温度从120-390 °C均有报道,但是过低的水热反应温度会在产物中留有Li3PO4,严重影响正极材料的电化学性能,而过高的合成温度对设备要求太高。本课题组以冷轧钢盐酸酸洗废液提取的Fe2+溶液为铁源,采用水热法制备LiFePO4正极材料,研究水热反应温度对正极材料的性能影响。实验结果表明,随着水热反应温度的提高,LiFePO4形貌趋于光滑的球形颗粒。他们认为反应温度的升高将导致水热体系的压力迅速增大,水热流体的粘度降低。而粘度的降低促进了水热体系中传质的进行,使得LiFePO4晶体的界面生长速率发生变化,因而形貌发生了转变。电化学性能测试中发现,随着反应温度的升高,正极材料的初始放电比容量先增大后减小。
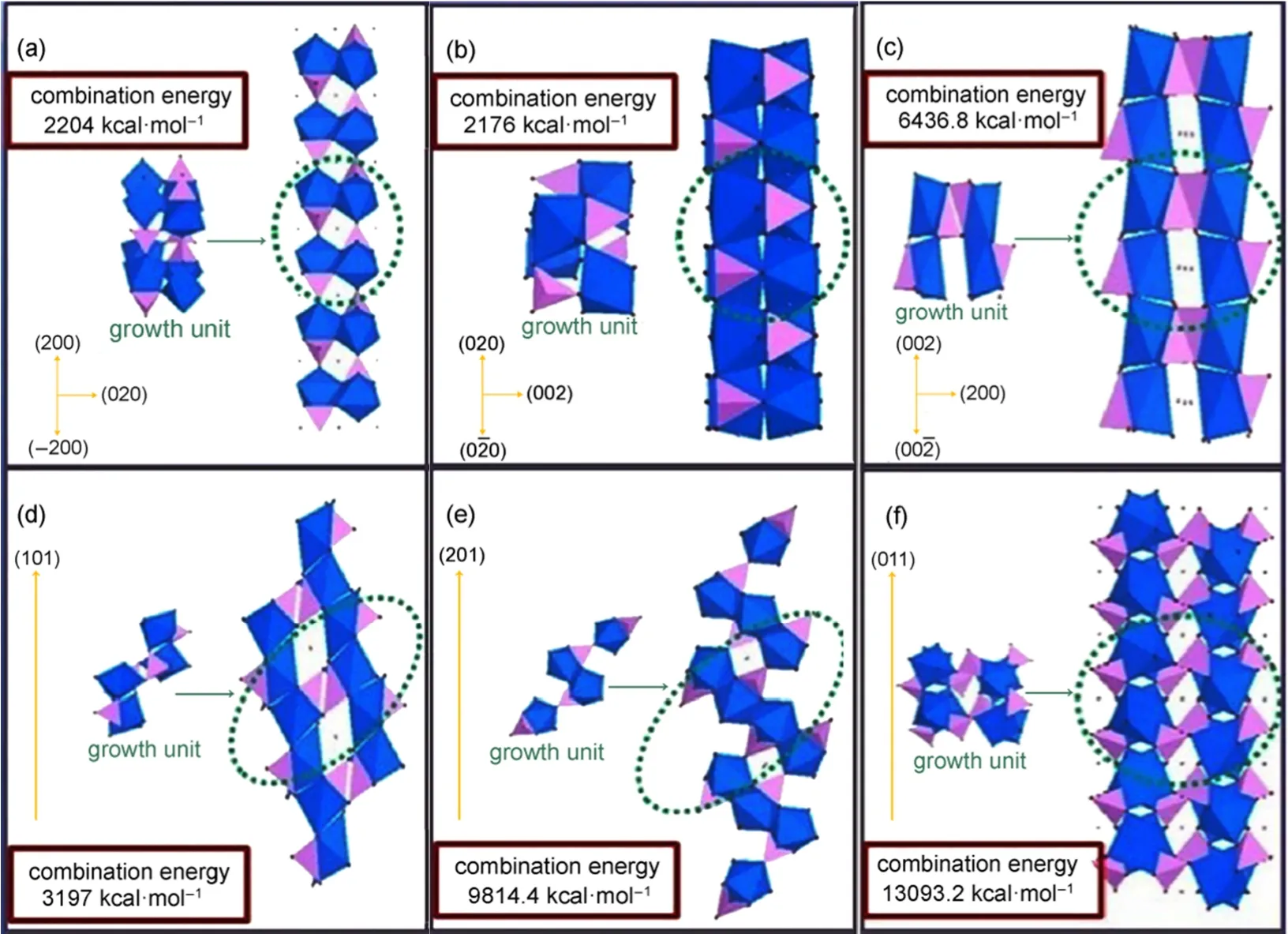
图10 不同晶面的有利生长单元及它们稳定组合的能量计算40Fig.10 Favorable growth units of various faces and the calculated stabilization energy of combination of growth units40
4.2 新型的制备方法
近年来,随着LiFePO4正极材料的深入研究,越来越多新型的LiFePO4制备方法开始频现,主要有:静电纺丝法42、燃烧法43、流变相法44、超临界法45、超声雾化热解法46、放电辅助机械球磨法47、溶液蒸干法48、气相合成法49、喷雾干燥法50、免模板反胶束法51、多元醇法52、冷冻干燥法53等。其中静电纺丝法、燃烧法、流变相法、超临界法等由于合成方法简易,制备正极材料的形貌可控,受到了科研工作者的广泛关注,因此本文主要就这几种方法做了简要综述。
4.2.1 静电纺丝法
静电纺丝法是聚合物溶液或熔体在静电作用下进行喷射拉伸而获得纳米级纤维的纺丝,作为一种新颖的纳米纤维制备方法。这种方法合成的复合材料通常具有大的比表面积和高的孔隙率,且电化学性能优异。同时,由于设备简单和过程安全可控等优点,是实现LiFePO4正极材料纳米化的一种新型制备方法54。
Shao等55以聚乙烯吡咯烷酮(PVP)用作静电纺丝模板和碳源,通过煅烧[LiOH·H2O + Fe(NO3)3·9H2O + H3PO4]/PVP电纺纳米带合成LiFePO4/C复合材料。LiFePO4/C复合纳米带的平均宽度为(2.50 ± 0.33) μm,平均厚度为约162 nm。电化学性能较好,在0.2C的电流密度下,初始放电比容量为123.38 mAh·g-1,50次循环后没有明显的容量衰减。Qiu等56以FeC6H5O7·5H2O和LiH2PO4为原料,聚乙烯吡咯烷酮(PVP)和聚环氧乙烷(PEO)作为共同的高聚物,通过传统的静电纺丝装置制备出LiFePO4/C纳米纤维。当LiH2PO4与FeC6H5O7·5H2O的摩尔比为1.3时,复合材料具有较高的相对容量、良好的纤维形态、粒径较小和良好的循环稳定性。在0.1C倍率下充放电,初始放电比容量达到116 mAh·g-1。Zhang等57采用静电纺丝法制备LiFePO4-碳纳米纤维复合材料,研究了复合纤维前驱体的热分解行为和热处理期间的反应,并探索了热处理参数对复合材料的微观结构、形态、碳含量、晶体结构和电化学性能的影响。实验结果表明,当复合纤维前驱体在空气中以2 °C·min-1的升温速率于280 °C下预处理4 h,然后在氩气气氛中以相同的升温速率于800 °C碳化14 h,得到的复合材料具有更高的初始放电比容量、更稳定的充放电循环性能和最佳的电化学性能。在0.5C的倍率下,初始放电比容量为146.3 mAh·g-1,100次循环后容量保持率较好。
4.2.2 燃烧法
燃烧合成,又称自蔓延高温合成,是利用反应物中金属、非金属的氧化还原置换反应所放出的巨大热量。与传统的合成方法相比,这种方法具有简单、快速、经济且产物纯度较高等优势,已用于制备氧化物(耐火氧化物、电介质、催化剂等)和非氧化物(碳化物、硼化物、硅化物、氮化物等)材料。由于它是一个高温过程,所以只能用来制备热力学稳定的材料。同时,快速加热和冷却的速率,使得亚稳态材料有可能获得新的和特殊的性质58。
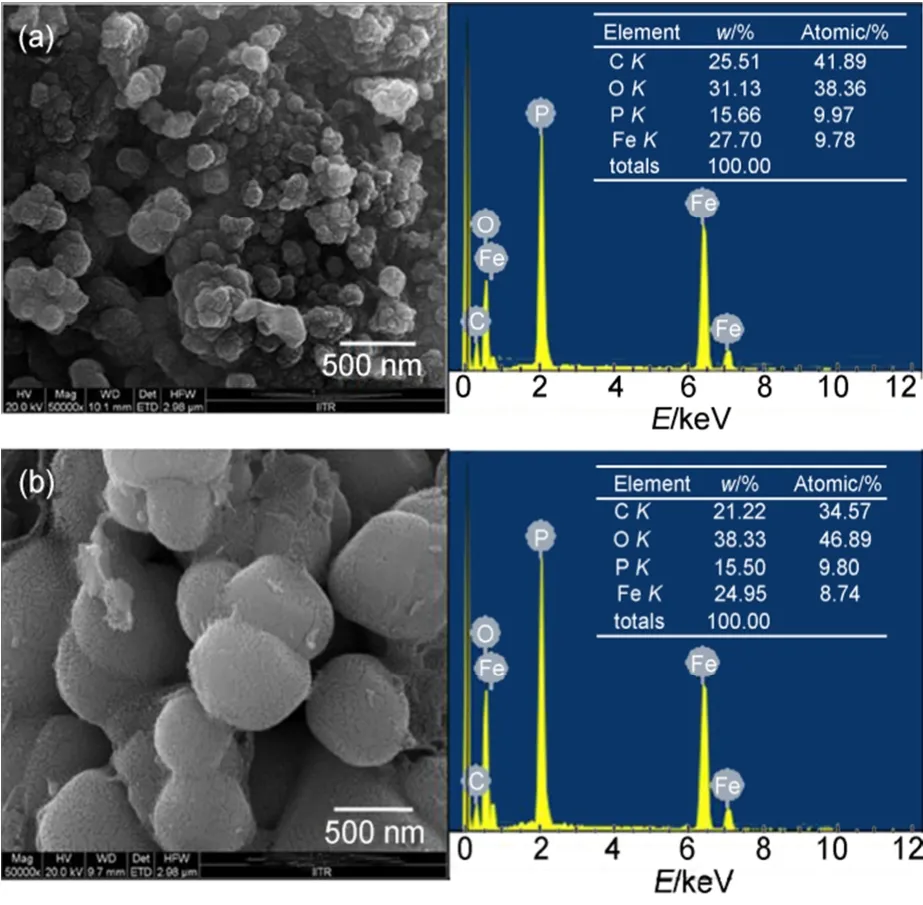
图11 LiFePO4(1.0)和LiFePO4(0.5)的FESEM图以及相关的EDAX光谱图59Fig.11 FESEM images of LiFePO4(1.0) and LiFePO4(0.5), EDAX spectra showing presence of C, O, P, and Fe59
Sehrawat等59通过聚合物凝胶燃烧法制备出LiFePO4/C复合材料,研究了添加不同量的苯胺单体形成凝胶对复合材料的性能影响。实验结果表明,以1.0 mL苯胺单体合成的LiFePO4(1.0)/C复合材料的颗粒尺寸大约为100 nm,表面碳涂层的厚度约为7 nm,均优异于0.5 mL苯胺单体合成的LiFePO4(0.5)/C复合材料,如图11和12所示。在快速放电倍率下的放电比容量较高,归因于复合材料较小的粒径、较低的电荷转移电阻和更高的锂离子扩散速率。Mohan等60以尿素为燃料,蔗糖作为碳源,采用燃烧法制备碳包覆的LiFePO4颗粒。实验结果表明,产物具有有序的橄榄石结构,平均微晶尺寸在30-40 nm的范围内,且具有良好的循环性能和倍率性能。在0.1C倍率下显示出较高的库仑效率,在0.5C和1C倍率下的比容量分别约144和134 mAh·g-1。但是,在高于1C倍率下的放电电压和可逆容量降低,主要是由于电极极化的加剧。Vujković等61采用凝胶燃烧制备掺杂V的LiFe1-xVxPO4/C复合材料。实验结果表明,产物的粒径尺寸较小、纯度较高。热处理过程中伴有磷化铁的形成,通过Rietveld分析确定其浓度。同时,V的引入不仅没有改变LiFePO4的晶体结构,还提高了复合材料的电化学性能。其中LiFe0.945V0.055PO4/C复合材料在饱和LiNO3溶液中以1C、10C和100C的倍率充放电,平均放电比容量分别为91、73和35 mAh·g-1,并显示出良好的循环性能。

图12 LiFePO4(1.0)和LiFePO4(0.5)的TEM及HRTEM图像59Fig.12 TEM and the corresponding HRTEM images of the LiFePO4(1.0) and LiFePO4(0.5)59
4.2.3 流变相法
流变相法是指将两种或两种以上的固体反应物经过混合研磨后,加入溶剂调制成流变态,放置于适当的反应条件以得到产物的方法。这种方法改变了其他传统制备工艺的繁杂过程,降低了合成温度,并缩短了煅烧时间。纯相产物颗粒粒径较小、热稳定性良好、电化学性能优异。同时,反应过程无固体排放物,对环境几乎无污染,属于绿色合成反应62。
Wu等63将自制FePO4·2H2O、LiOH·H2O、聚乙二醇和适量的水混合并研磨,得到固-液流变体,在管式炉中煅烧流变体,获得LiFePO4/C复合材料。同时,研究了不同煅烧温度对材料的电化学性能的影响。实验结果表明,当煅烧温度为650 °C时,复合材料显示出高容量和良好的循环性,在1C倍率下的初始放电比容量为153.2 mAh·g-1,50次循环后放电比容量仍达到151.3 mAh·g-1,容量保持率高达98.8%,归因于较高煅烧温度会导致材料更大的粒度,增加锂离子扩散路径,从而影响复合材料的电化学性能。Tang等64采用流变相法制备LiFePO4正极材料,研究了包覆导电材料Li0.34La0.51TiO2.94(LLTO)对正极材料的晶体结构和电化学性能的影响。实验结果表明,LLTO纳米涂层不会影响LiFePO4正极材料的橄榄石型结构。电化学性能测试中,室温下LLTO-LiFePO4复合材料在0.1C和1C倍率下的初始放电比容量分别为158.1 mAh·g-1和148.3 mAh·g-1,在10C高倍率下的放电比容量达到109.9 mAh·g-1,20次循环后比容量仅衰减3.2%。优异的倍率性能和循环性能归因于流变相法制备的LLTO-LiFePO4复合材料具有较小的颗粒尺寸,减小了锂离子扩散路径的距离,且LLTO纳米涂层阻碍了LiFePO4颗粒与电解质直接接触,减少电解液对LiFePO4的侵蚀。Li等65以乙酸作为分散剂,采用流变相法制备掺杂Mg的LiFePO4/C(LMFP)复合材料,通过研究煅烧参数、Li/Fe值、Mg掺杂量和涂层碳含量对复合材料的形貌和电化学性能的影响。得到的最佳复合材料显示出优异的倍率性能和循环稳定性能,在5C倍率下的初始放电比容量为141 mAh·g-1,在20C倍率下为111 mAh·g-1,且100次循环后具有99.1%的容量保持率,如图13所示为LMFP与未掺杂的LiFePO4/C(LFP)复合材料的电化学性能图。
4.2.4 超临界法
当温度和压力同时增大到一定数值时,气液两相线到达终点,物质达到一个新的状态,即超临界状态。温度和压力处于临界压力和临界温度之上的流体称为超临界流体。超临界流体同时具有气体和液体的特征,有类似于气体的粘度,同时具有与液体相当的密度66,67。在超临界流体中,反应速率和成核速率提升,产物的结晶度高、粒径小。通过调控状态参数,可以调节产物的形貌和颗粒尺寸68。
张艳洁等69采用超临界水热法制备具有高纯度和结晶度的LiFePO4正极材料,研究了溶剂和煅烧等工艺参数对产物的纯度、尺寸和形貌的影响。实验结果表明,水和乙醇的混合溶剂有利于合成更小、更均匀的颗粒,以400 °C高温煅烧,产物的尺寸为约100-300 nm。测试电化学性能中,在0.1C和1C倍率下的初始放电比容量分别为151.3和128.0 mAh·g-1,在1C倍率下100次循环后的容量保持率为95.0%。Rangappa等70以油胺作为封端剂和还原剂,采用超临界乙醇法制备LiFePO4纳米颗粒,研究了不同反应条件对LiFePO4纳米颗粒的尺寸、形貌和电化学性能的影响。实验结果表明,油胺与乙醇的较高体积比(> 1 : 1)、较短的反应时间(4 min)和较低的反应温度(300-320 °C)有利于形成粒径尺寸小于20 nm的片状结构LiFePO4纳米颗粒。他们将此现象归因于超临界条件下过饱和度和形核速率的提高促进了LiFePO4纳米颗粒的形核和生长过程的分离,从而形成小而窄的粒径分布。油胺覆盖在LiFePO4纳米颗粒的表面,抑制进一步的颗粒生长。同时,超临界乙醇较低的介电常数也发挥了关键作用。测试电化学性能中,复合材料在0.1C倍率下的初始放电比容量为158 mAh·g-1,且循环性能优异。此外,Xie等71,72通过引入超临界二氧化碳改变LiFePO4颗粒的形态,研究了多孔LiFePO4的转化机理。实验结果表明,超临界二氧化碳将聚集的LiFePO4片晶分散成尺寸为约3-5 mm菱形板块,并形成多孔LiFePO4。他们认为这些多孔结构起源于超临界二氧化碳的溶解在LiFePO4表面形成的凹坑。多孔结构仅发生在菱形LiFePO4板的侧面上,这是由于LiFePO4晶体结构的各向异性。电化学性能测试发现,多孔结构大大提高了LiFePO4的倍率性能,在0.1C倍率下的放电比容量为162 mAh·g-1,且容量保持率优异。
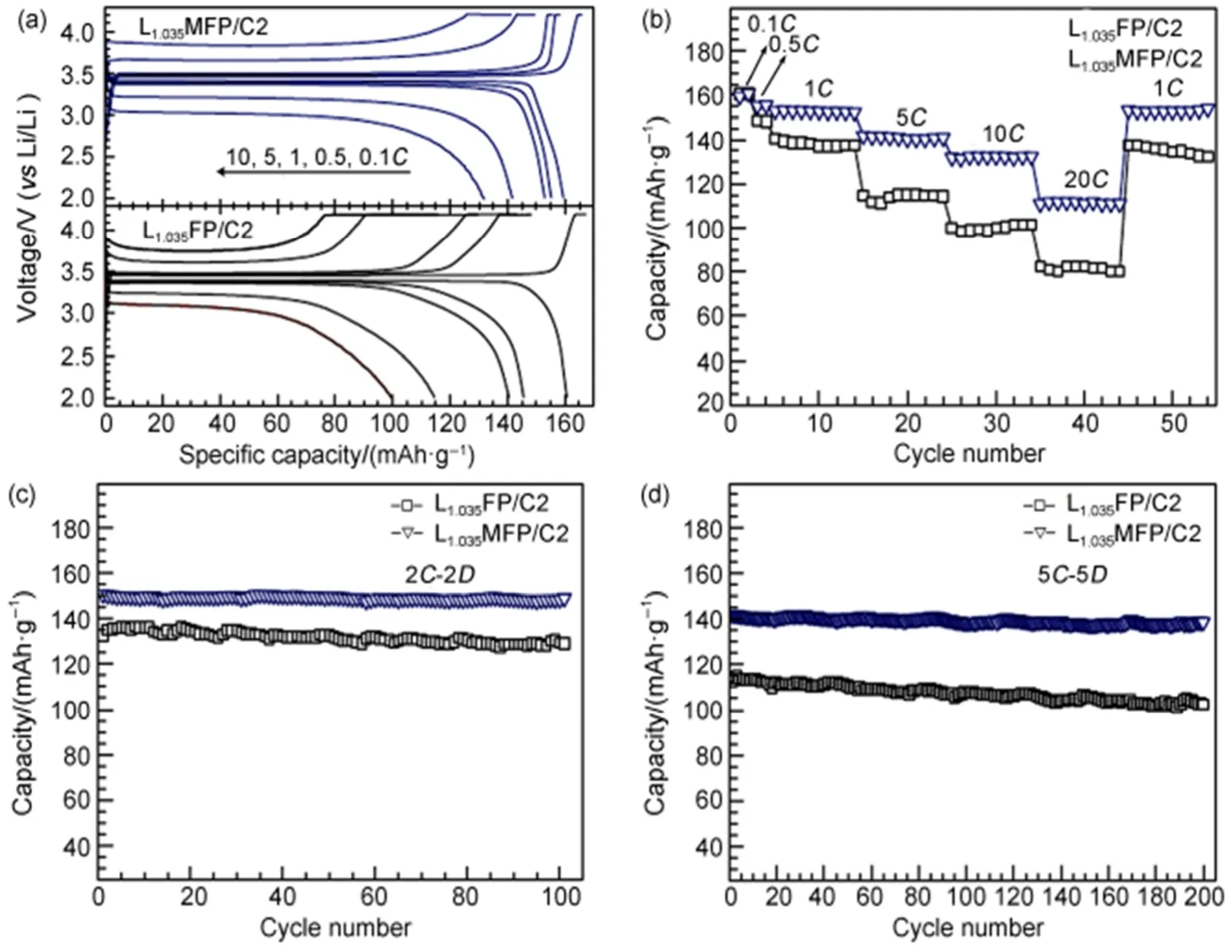
图13 Mg掺杂和未掺杂的LiFePO4/C复合材料的电化学性能图65Fig.13 Electrochemical performance of Mg-doped and undoped LiFeP O4/C composite65
5 LiFePO4的改性研究
LiFePO4作为最具潜力的锂离子电池正极材料,本身也存在着一些缺陷,主要是电导率和锂离子扩散速率较低,从而限制了其广泛应用。针对这些问题,目前主要有三种解决方法73,74:一是向LiFePO4晶格中掺杂离子,提高其电导率和锂离子扩散速率75,76;二是添加导电剂及表面改性剂,如碳包覆、金属基包覆等,提高正极材料的导电性能77-80;三是细化LiFePO4晶粒,控制粒径尺寸,从而提升正极材料得电化学性能81,82。
5.1 离子掺杂改性
掺杂改性是使掺杂离子进入LiFePO4晶格内部取代部分离子的位置,以提高锂离子扩散速率和导电性能,降低电池内阻。掺杂改性包括金属离子掺杂和非金属离子掺杂。未掺杂和掺杂的LiFePO4正极材料分别表现为n型半导体和p型半导体,金属离子的掺入使得p型半导体载流子增加,以提高正极材料的整体导电性。掺杂后的充放电循环过程中,正极材料中的Fe呈现混合价态,且Fe3+/Fe2+的比例发生了改变,从而导致了正极材料在p型和n型两种型态之间转变,极大地增加LiFePO4正极材料的导电性。而电负性较大的阴离子掺杂可以增加电池内部电势差,提高正极材料的充放电比容量83。但是,不是所有的离子都适合作为掺杂离子,在选择掺杂离子时应考虑一定的影响因素,主要为两个方面:(1) 掺杂离子的化合价:掺杂离子的化合价态高,在晶格中形成的空穴就越多,有利于离子的通过,提高锂离子在正极材料中的扩散速率;(2) 掺杂离子的半径:掺杂离子半径应与所替代的离子半径相接近,这样比较容易进入所替代离子的位置84。表1为不同离子掺杂改性后,LiFePO4正极材料性能研究的总结。

表1 掺杂不同离子对LiFePO4的改性作用Table 1 Modification effect of doping with different ions on LiFePO4
离子掺杂改性时,一般采用LiFePO4晶格中的Li位、Fe位或O位的掺杂,P位的掺杂较少。由于Li位的掺杂不利于Li+的扩散,可供选取的掺杂元素较少,掺杂效果较好的元素有Na、Mg、Ta等;Fe位的掺杂可以削弱Li―O键的相互作用,提高离子的流动性和扩散系数,改善LiFePO4正极材料电化学性能较明显的元素有Sn、Mg、Cu、Cr、Mn等;O位的掺杂元素有F、Cl等105-107。自2002年Chung等102采用掺杂金属离子使LiFePO4正极材料的电导率提高了8个数量级后,研究人员开始对掺杂改性技术进行更深入的研究。现如今,经过离子掺杂后的LiFePO4正极材料的电导率可以提高到3 × 10-3-4 × 10-2S·cm-1,超过传统正极材料LiCoO2(~10-4S·cm-1)和LiMn2O4(~10-6S·cm-1)的电导率108。但是,离子掺杂改性的研究还存在一些问题:一是目前的掺杂改性研究主要集中在选取离子的种类方面,对不同位的掺杂及其机理研究较少,如何实现离子的有效掺杂以及其机理如何需要进一步探讨;二是对离子掺杂的效果评价还存在不同观点,有研究人员认为是煅烧过程中前驱体分解的碳被遗留下来,致使正极材料的电导率提高,也有Zaghib109、Harrison110、Herle111、Delacourt112等认为掺杂的离子没有进入LiFePO4晶格内部,而是生成的具有贯通纳米相网络结构的金属磷化物提高了正极材料的电导率。这些问题说明离子掺杂改性的研究仍需进一步深入。

continued Table 1
5.2 包覆改性
在LiFePO4正极材料的表面上包覆导电物质是改善其电化学性能的重要方法之一。导电物质的包覆一方面可以增强粒子之间的导电性,另一方面也为LiFePO4晶体提供电子隧道,补偿锂离子脱/嵌过程中的缺失电荷数。常用的包覆改性方法包括:碳包覆、金属基包覆、金属氧化物包覆、固体电解质包覆和导电聚合物包覆等。
5.2.1 碳包覆
碳包覆实质上是在LiFePO4颗粒表面包覆了一层导电性能较好的碳膜,既可以增强材料的电导性,补偿脱/嵌过程中锂离子的电荷平衡。同时,由于碳的还原作用,还可以防止材料中Fe2+被氧化,有效阻止颗粒间的团聚,而碳并未进入材料晶格113。Ravet等114于1999年最先报道了在LiFePO4颗粒表面的碳包覆改性,当正极材料包覆1% (w)的碳涂层后,在80 °C下以1C倍率充放电,获得的放电比容量接近理论数值,该结果表明了碳包覆是提高LiFePO4正极材料比容量的有效方法。但是,碳包覆需要经过高温处理,存在LiFePO4颗粒的粒径增大、颗粒只有部分包覆碳等问题。近年来,对碳包覆的研究报道越来越多,研究工作者通过选取不同种类的碳包覆材料,控制包覆碳的含量,改进合成工艺,可以将正极材料的电导率提高6-8个数量级,达到1.7 × 10-3S·cm-1,已渐渐解决上述问题115。目前常被使用的碳包覆材料包括:蔗糖116、葡萄糖117、乙炔黑118、石墨烯119、抗坏血酸120、柠檬酸121、多壁碳纳米管122、聚丙烯123、聚乙烯醇124等。
Gong等17概述了碳源为一维纳米碳、二维纳米碳和三维纳米碳等先进碳材料的结构和制备方法,并对LiFePO4包覆先进碳材料后的电化学性能及其应用进行了详细的讨论。研究表明,先进碳材料由于电子电导率优越和具有较高的比表面积,使得复合材料具有优良的电化学性能,特别是高倍率性能和超循环寿命。Du等125以原位水热法合成LiFePO4/石墨烯复合材料,并对复合材料的3D结构进行了系统的研究。研究表明,3D结构提供了导电网络,由此提高了复合材料的导电性和电子传输速率。同时,石墨烯的包覆有利于减小LiFePO4纳米颗粒的尺寸,这也是性能得以优化的另一个原因。进行电化学性能测试时,复合材料在0.2C倍率下的放电比容量为160 mAh·g-1,10C倍率下达到115 mAh·g-1,100次循环后容量保持率为94.2%,表现出良好的倍率性能和循环性能。此外,采用传统合成方法制备出的LiFePO4/氧化石墨烯复合材料,由于LiFePO4的二维特征,氧化石墨烯只有点接触材料,而不能发挥其全部效果。Li等126提供了在一种氧化石墨烯上生长FePO4粒子从而获得LiFePO4前驱体的方法,负电荷氧化石墨烯片可以促进FePO4颗粒的生长。以自制氧化石墨烯为原料,采用沉淀法制备出FePO4·xH2O/氧化石墨烯复合材料,再以碳热还原法制备出了LiFePO4/氧化石墨烯复合材料。所得复合材料的颗粒粒径较小,尺寸范围从50-300 nm,具有较好的电化学性能,在0.1C倍率下的初始放电比容量为163.4 mAh·g-1。
5.2.2 金属基包覆
由于金属离子的电导性较好,可用作LiFePO4正极材料的金属基包覆改性。卢俊彪等127以自制Fe(CO2)2·2H2O、Li2CO3、NH4H2PO4和AgNO3为原料,聚乙二醇为分散剂,无水乙醇为介质,利用球磨工艺制备LiFePO4/Ag复合材料,研究了电流密度的大小与充放电比容量、电极极化效应之间的关联。测试电化学性能中,在C/8的倍率下45次循环后,复合材料的充放电比容量在160-165 mAh·g-1,且具有较好的稳定性能。Yang等128通过X射线衍射、拉曼光谱和电化学阻抗光谱系统地研究了由纳米Si包覆改性后的LiFePO4/(C+Si)复合材料和LiFePO4/C复合材料的Li+动力学过程。研究表明,LiFePO4/(C+Si)复合材料的充放电速率比LiFePO4/C复合材料的更为优异,前者具有更大的锂离子扩散速率。同时,纳米Si的加入有效抑制Fe溶解,提高复合材料的稳定性能和循环性能。Saliman等129在LiFePO4/C复合材料表面上沉积Pd纳米颗粒。研究表明,Pd纳米颗粒聚集在材料表面,且聚集体(次级纳米颗粒)呈球形,尺寸为约20-40 nm。在10C高倍率下的初始放电比容量达到98 mAh·g-1,说明Pd纳米颗粒具有良好的导电性,提高了复合材料的电化学性能。
5.2.3 金属氧化物包覆
Zhao等130通过采用溶胶-凝胶法在商业LiFePO4/C复合材料的表面上的制备纳米Al2O3涂层,研究了Al2O3涂层对室温和高温下LiFePO4的电化学性能和结构稳定性的影响。研究表明,2% (w)的Al2O3涂层可以有效提高循环能力,缓解高温下的容量衰减,降低电池阻抗。Xu等131采用水热法合成LiFePO4原料,并以溶胶-凝胶法在LiFePO4的表面制备碳和TiO2混合涂层。研究表明,混合涂层中适量的TiO2可以显着提高LiFePO4的倍率性能,这是由于TiO2颗粒扩大了涂层中的界面,混合结构可以为Li+提供更多脱/嵌的反应位点。同时,这种碳和TiO2混合涂层的另一个优点是其不需要严格限制涂层中的碳含量,有益于LiFePO4碳涂层工艺的规模生产。除了使用单一的金属氧化物包覆,Tang等132以铝掺杂氧化锌(AZO)为包覆层,通过高温固相法制备倍率、循环性能优异的LiFePO4正极材料。研究表明,AZO均匀包覆在样品表面,既改善了复合材料的倍率性能和低温性能,又增加了复合材料的振实密度。用作锂离子正极材料时,室温下以20C高倍率充放电,放电比容量达到100.9 mAh·g-1,在-20 °C时进行0.2C倍率下充放电,复合材料的放电比容量为119.4 mAh·g-1,归因于AZO包覆增加了复合材料的电导率,从而提高了其比容量。
5.2.4 固体电解质包覆
张晓萍等133采用机械活化技术制备了性能优异的9LiFePO4/Li3V2(PO4)3复合材料。研究表明,部分V3+进入LiFePO4晶格内部,使其晶格参数减小,复合材料的交换电流密度和锂离子扩散系数均提高了1个数量级,且电化学性能得到提升。Ma等134用溶胶-凝胶燃烧法制备用LiFePO4/(C+ Li3V2(PO4)3)(LFP/C-LVP)复合材料。研究表明,复合材料的电化学性能优异,在1C、2C、5C和10C倍率下的放电比容量分别为153.1、137.7、113.6和93.3 mAh·g-1,且低温性能较好。同时,证明了Li3V2(PO4)3的存在使得复合材料在-20 °C至25 °C低的范围内电荷转移电阻降低,增强了电化学催化活性,如表2所示。此外,Ma等分别以CePO4135和LaPO4136包覆LiFePO4正极材料。研究表明,在样品表明沉积合适量的CePO4和LaPO4都显示出更高的可逆容量和稳定的循环性能,归因于涂层良好的导电性和稳定性,增强了复合材料表面上的电子和锂离子传输,提高了电极的转移动力学。LiFePO4/(C+CePO4)复合材料在1C、2C、5C和10C倍率下的初始放电比容量分别为166.1、161.4、143.7和120.3 mAh·g-1,即使在10C倍率下650次循环后,容量保持率仍保持高达77.5%。此外,复合材料在-20 °C下显示出100.9 mAh·g-1的较高可逆容量。而LiFePO4/(C+LaPO4)复合材料在1C、2C、5C和10C倍率下的初始放电比容量分别为150.7、142.3、116.6和80.3 mAh·g-1,在5C和10C倍率下循环100次后,可逆容量分别为115.1和79.3 mAh·g-1,保持率高达98.5%和98.3%。

表2 不同温度下电化学阻抗和交换电流密度的结果134Table 2 Result of electrochemical impedance and exchange current density at various temperatures134
5.2.5 导电聚合物包覆
近年来,对LiFePO4正极材料的包覆改性除了上述的四种方法外,基于碳包覆衍生出的导电聚合物包覆改性也成为专家、学者研究的热点。与碳包覆不同,导电聚合物包覆通常要经过单体的氧化聚合,形成网状或三维结构,以此来提高正极材料的电导率和锂离子扩散速率,达到优化正极材料的倍率性能、低温特性和循环稳定性的效果。
Bai等137采用原位聚合噻吩单体,在LiFePO4颗粒表面包覆聚噻吩(PTh),获得LiFePO4/PTh复合材料。研究表明,PTh涂层降低了LiFePO4正极材料的电荷转移电阻,提高了复合材料的电化学性能,使得复合材料的可逆容量和循环性能都有所提升。Huang等138以聚吡咯(PPy)和聚苯胺(PANI)作为导电聚合物,均采用电化学沉积和化学聚合两种方法制备复合材料,但由于前驱体溶液中酸的存在会破坏LiFePO4颗粒的核-壳结构,导致不能通过电化学沉积获得LiFePO4/(C+PANI)复合材料。研究表明,两种导电聚合物的包覆均使复合材料的电化学性能提升。采用化学聚合制备的具有7% (w) PPy或PANI的复合材料均表现出最佳的电化学性能,而采用电化学沉积制备的具有20% (w) PPy的LiFePO4/(C+PPy)复合材料的电化学性能最佳,他们认为电化学沉积制备复合材料时需要更多的PPy作为导电物和粘合剂,而对于化学聚合制备的复合材料,不仅添加了PPy或PANI,还需要有炭黑和PTFE的加入。最近,Sehrawat等59也研究了包覆不同含量的PANI对LiFePO4/ (C+PANI)复合材料电化学性能的影响。实验结果表明,1.0 mL苯胺单体制备的LiFePO4/(C+PANI)复合材料在5C和10C高倍率下的放电容量为72和60 mAh·g-1,优于0.5 mL苯胺单体制备的复合材料。电化学性能的改善归因于导电聚合物的包覆导致复合材料粒径尺寸的减小、较低电荷转移阻抗和更高的锂离子扩散速率。除了包覆PANI材料,Sehrawat等139制备的LiFePO4/(C+PPy)复合材料同样表现出优异的电化学性。此外,他们还深入探讨了溶剂对合成PPy的性质的影响。研究表明,以乙腈为溶剂合成的PPy用于正极材料的包覆,其电化学性能最佳,归因于导电聚合物涂层的形态和结构。

图14 粒径对LiFePO4材料的放电比容量的影响146Figure 14 Effects of particle size on the specific discharge capacity of LiFePO4materials146
5.3 晶粒细化
2001年,Yamada等140通过改进合成工艺参数,制备出粒径较小的LiFePO4颗粒,室温下在0.12 mA·cm-2电流密度下的放电比容量达到理论数值的95%。由此,晶粒细化便成为提高LiFePO4正极材料电化学性能的另一种有效方式。晶粒细化,实际上就是缩短了锂离子和电子的扩散路径,从而增加正极材料的导电性能。同时,较小的颗粒会降低晶体的缺陷密度。但是,晶粒纳米化也会增加LiFePO4颗粒的表面能,导致其稳定性降低,且晶粒纳米化通常需要在低温下进行,而较低温度会导致正极材料的结晶度较差,降低其电化学性能141。结合碳包覆和晶粒细化的思想,Huang等142制备了粒径为100-200 nm的LiFePO4颗粒,并包覆大约15% (w)的碳。即使在5C倍率下充放电,复合材料的放电比容量达到120 mAh·g-1,首次证明LiFePO4正极材料在室温下具有优异的倍率性能。多年来,研究工作者也致力于解决晶粒细化所带来的一些问题,如今已有了显著成效。
Saravanan等143采用溶剂热法制备具有不同厚度的LiFePO4/C纳米板,研究了合成工艺对LiFePO4/C纳米板形态的影响。研究表明,不同铁源制备出的LiFePO4/C纳米板分别呈分层状、纺锤状、厚板状、金刚石状等形态,厚度在20-500 nm之间。进行电化学性能测试,厚度约为30 nm的分层状LiFePO4/C纳米板表现最为优异,在0.1C倍率下的初始放电比容量达到167 mAh·g-1,30C高倍率下的初始放电比容量为46 mAh·g-1。此外,他们发现随着LiFePO4/C纳米板厚度的减小,其电化学性能将提高,但厚度过小会造成团聚现象,导致LiFePO4/C纳米板的电化学性能下降。Hong等144采用连续超临界水热法制备LiFePO4/C复合材料,研究了流体的流速和混合器的几何形状对合成复合材料性质的影响。研究发现,随着流体的流速增大,LiFePO4颗粒的粒径减小,但流速过快会导致形成Fe3+杂质的增加,使得复合材料的放电比容量下降。同时,选取适当的流体流速,使用旋涡式三通混合器合成的LiFePO4颗粒的粒径为100-400 nm。室温下在0.1C倍率的初始放电比容量为149 mAh·g-1,55 °C高温下在20 C倍率的初始放电比容量也达到85 mAh·g-1,优于90°式和50°式三通混合器合成的LiFePO4/C复合材料,他们归因于前者复合材料的较高结晶度和较少杂质含量。Liu等145采用流变相结合碳热还原工艺制备LiFePO4/C复合材料,研究了前驱体粒径对产物颗粒尺寸的影响,并比较了不同晶粒尺寸的LiFePO4/C复合材料之间的性能。研究表明,采用高能球磨工艺可将前驱体的粒径减小到95 nm,高温煅烧后形成40-100 nm的单晶LiFePO4/C纳米复合材料,且比表面积高达48.0 m2·g-1,均优于未经处理的前驱体合成的LiFePO4/C复合材料。此外,单晶LiFePO4/C纳米复合材料的循环伏安曲线显示更好的可逆反应性能和较低的极化,在10C倍率下的初始放电比容量也高达100 mAh·g-1,循环1000次后的容量保持率达到90%。他们将单晶LiFePO4/C纳米复合材料表现出的优异性能归因于碳包覆和晶粒细化起到的重要作用。原位碳包覆增强了电子传导性,并限制晶粒生长和抑制晶粒团聚,从而使纳米尺寸的前驱体在煅烧过程中更容易形成单晶颗粒。同时,晶粒细化不仅减小了离子和电子传输的距离,单晶纳米复合材料的比表面积也得以增大。值得注意的是,采用碳热还原方法制备出单晶结构的LiFePO4鲜有报道,他们的研究将为晶粒细化的进一步研究提供新思路。最近,Liu等146研究了未涂覆的LiFePO4颗粒的晶粒尺寸与电化学性能之间的关系。为了获取粒度分布均匀的产物,他们通过控制合成工艺参数,采用共沉淀法制备出了具有30-500 nm的不同晶粒尺寸的LiFePO4颗粒。研究表明,较高的回流温度增强了LiFePO4颗粒的结晶度,而较长的回流时间可以使杂质相的形成最小化。400 °C是形成良好结晶的LiFePO4颗粒的最低煅烧温度。同时,在不受包覆碳影响的情况下,纯LiFePO4正极材料的放电比容量与不同晶粒尺寸呈“火山”型关系,如图14所示,“火山”曲线的顶部位于约200 nm的颗粒尺寸。这一研究可能推动LiFePO4正极材料向更精确的晶粒细化方向发展。
6 总 结
充放电过程中,随着LiFePO4正极材料中锂离子的嵌入和脱出,往往带来了正极材料晶体结构的显著变化,而这些微观变化与正极材料的电化学反应机理、充放电比容量值、电压曲线、体积变化等密切相关。研究人员通过获得LiFePO4正极材料在充放电过程中的相组成及相结构演化,解释了LiFePO4的充放电行为,是LiFePO4正极材料基础研究的核心内容,对改善其物理化学及电化性能具有十分重要的意义。
合成方法是制备纯相LiFePO4的关键,直接影响到正极材料的性能,并决定着其是否可以实现商业化扩大生产。传统的合成方法已相对较为成熟,并被广泛运用,但技术上仍然存在一些问题,诸如产物颗粒不均匀、合成周期较长、对设备要求较高等,阻碍了LiFePO4正极材料的快速发展。静电纺丝法、燃烧法、流变法、超临界法等新型合成方法的出现,不仅拓宽了制备LiFePO4正极材料的途径,而且新型方法对设备、技术和环境的要求简便,合成的产物性能更优异。
由于LiFePO4正极材料的电导率和锂离子扩散速率较低,以其进行商业化发展仍存在巨大的挑战147。为了改善LiFePO4正极材料的导电性能,研究人员已经尝试了对其结构和微结构的多样改性,如离子掺杂改性、包覆改性和晶粒细化。离子掺杂改性,就是通过引入离子代替LiFePO4晶格内部部分离子的位置,使正极材料产生晶格缺陷,促进Li+扩散,从而改善晶体内部的导电性能。使用较多的离子包括:Nb5+、Ti4+、Al3+、Mg2+、F-等。掺杂离子的化合价态越高,在晶格中形成的空穴就越多,提高锂离子的扩散速率。同时,掺杂离子的半径与所替代离子的半径越接近,越容易进入所替代离子的位置,从而提高导电性能,降低电池内阻。但是,关于掺杂离子提高LiFePO4正极材料电导率的机理还未定论,有待进一步深入探究。包覆改性,就是在LiFePO4颗粒表面包覆或分散适当的导电材料,增强了粒子与粒子之间的导电性,但不会改变正极材料的结构,又可以抑制颗粒的长大。目前,碳包覆改性技术相对较优,虽然包覆碳会降低正极材料的振实密度,但包覆的碳涂层可以起到还原的作用,避免Fe2+的氧化。此外,还有银、铜等金属物的包覆,也可以优化LiFePO4正极材料的电化学性能,但是成本比较高,不适用于工业化大规模的生产。因此,导电材料的选择和添加量必须明确,在提高正极材料性能的同时,需要降低制备成本148,149。晶粒细化,就是减小LiFePO4颗粒的粒径尺寸,通过改进合成工艺参数得以实现。晶粒纳米化使得锂离子和电子的扩散路径缩短,比表面积增大,晶体的缺陷密度降低,但是过小的晶粒粒径容易产生团聚现象。因此,需合成适当大小的粒径尺寸,提升LiFePO4正极材料的电化学性能。
近年来,通过优化制备工艺,以掺杂、包覆、晶粒细化等技术制备出了性能优异的LiFePO4正极材料,在低倍率下的初始放电比容量可达169.9 mAh·g-1,非常接近理论数值,较长周期循环后的容量保持率仍可维持在90%以上,且-20 °C下的放电容比量也达到147 mAh·g-1。此外,在10C高倍率下的初始放电比容量高达129.8 mAh·g-1。循环性能、低温特性和高倍率充放电性能得到了大幅度的提升。同时,随着产业化技术的不断优化,未来LiFePO4电池除了在电动工具、自行车和代步车市场外,在航空航天、电动汽车和混合动力电动汽车等领域也将有着广泛的应用前景。
7 展 望
数十年来,国内外专家、学者对LiFePO4正极材料的研究已有了显著成效,但受正极材料理论比容量的限制和结构稳定性的制约,研究人员不可能只依靠改进现有的工艺技术实现材料根本性突破。由于在锂离子电池充放电过程中,正极材料的结构变化与其电化学反应机理和性能密切相关,未来研究可以通过在原子尺度上原位观察LiFePO4正极材料充放电过程中脱/嵌锂前后的微观结构,从更深层次认识其电化学反应机理和性能演化规律,从而全面了解LiFePO4正极材料的电化学行为,为其规模化应用提供新思路。
(1) Padhi, A. K.; Nanjundaswamy, K. S.; Goodenough, J. B. J. Electrochem. Soc. 1997, 144 (4), 1188. doi: 10.1149/1.1837571
(2) Padhi, A. K.; Nanjundaswamy, K. S.; Masquelier, C.; Okada, S.; Goodenough, J. B. J. Electrochem. Soc. 1997, 144 (5), 1609. doi: 10.1149/1.1837649
(3) Bi, Z. Y.; Zhang, X. D.; He, W.; Min, D. D.; Zhang, W. S. RSC Adv. 2013, 3 (43), 19744. doi: 10.1039/C3RA42601G
(4) Dimesso, L.; Förster, C.; Jaegermann, W.; Khanderi, J. P.; Tempel, H.; Popp, A.; Engstler, J.; Schneider, J. J.; Sarapulova, A.; Mikhailova, D.; Schmitt, L. A.; Oswaldc, S.; Ehrenbergd, H. Chem. Soc. Rev. 2012, 41 (15), 5068. doi: 10.1039/C2CS15320C
(5) Sun, X. F; Xu, Y. L.; Liu, Y. H.; Li, L. Acta Phys. -Chim. Sin. 2012, 28 (12), 2885. [孙孝飞, 徐友龙, 刘养浩, 李 璐. 物理化学学报, 2012, 28 (12), 2885.] doi: 10.3866/PKU.WHXB201209271
(6) Zhang, Y.; Huo, Q. Y.; Du, P. P.; Wang, L. Z.; Zhang, A. Q.; Song, Y. H.; Lv, Y.; Li, G. Y. Synth. Met. 2012, 162 (13), 1315. doi: 10.1016/j.synthmet.2012.04.025
(7) Lu, L. G.; Han, X. B.; Li, J. Q.; Hua, J. F.; Ouyang, M. G. J. Power Sources 2013, 226 (3), 272. doi: 10.1016/j.jpowsour.2012.10.060
(8) Amin, R.; Balaya, P.; Maier, J. Electrochem. Solid-State Lett. 2007, 10 (1), A13. doi: 10.1149/1.2388240
(9) Morgan, D.; Ven, A. V. D.; Ceder, G. Electrochem. Solid-State Lett. 2004, 7 (2), A30. doi: 10.1149/1.1633511
(10) Chung, S. Y.; Chiang, Y. M. Electrochem. Solid-State Lett. 2003, 6 (12), A278. doi: 10.1149/1.1621289
(11) Xu, Y. N.; Chung, S. Y.; Bloking, J. T.; Chiang, Y. M.; Ching, W. Y. Electrochem. Solid-State Lett. 2004, 7 (6), A131. doi: 10.1149/1.1703470
(12) Jugović, D.; Uskoković, D. J. Power Sources 2009, 190 (2), 538. doi: 10.1016/j.jpowsour.2009.01.074
(13) Andersson, A. S.; Thomas, J. O.; Kalska, B.; Häggström, L. Electrochem. Solid-State Lett. 2000, 3 (2), 66. doi: 10.1149/1.1390960
(14) Lv, W. Q.; Niu, Y. H.; Jian, X.; Zhang, K. H. L.; Wang, W.; Zhao, J. Y.; Wang, Z. M.; Yang, W. Q.; He, W. D. Appl. Phys. Lett. 2016, 108 (8), 1188. doi: 10.1063/1.4942849
(15) Abdellahi, A.; Akyildiz, O.; Malik, R.; Thorntonc, K.; Ceder, G. J. Mater. Chem. A. 2016, 4 (15), 5436. doi: 10.1039/C5TA10498J
(16) Masrour, R.; Hlil, E. K.; Obbade, S.; Rossignol, C. Solid State Ionics 2016, 289, 214. doi: 10.1016/j.ssi.2016.03.016
(17) Gong, C. L.; Xue, Z. G.; Wen, S.; Ye, Y. S.; Xie, X. L. J. Power Sources 2016, 318 (30), 93. doi: 10.1016/j.jpowsour.2016.04.008
(18) Bruce, P. G. Chem. Commun. 1997, 19 (19), 1817. doi: 10.1039/A608551B
(19) Yuan, L. X; Wang, Z. H.; Zhang, W. X.; Hu, X. L.; Chen, J. Tao.; Huang, Y. H.; Goodenough, J. B. Energy Environ. Sci. 2011, 4 (2), 269. doi: 10.1039/C0EE00029A
(20) Srinivasan, V.; Newman, J. J. Electrochem. Soc. 2004, 151 (10), A1517. doi: 10.1149/1.1785012
(21) Laffont, L.; Delacourt, C.; Gibot, P.; Wu, M. Y.; Kooyman, P.; Masquelier. C.; Tarascon, J. M. Chem. Mater. 2006, 18 (23), 5520. doi: 10.1021/cm0617182
(22) Delmas, C.; Maccario, M.; Croguennec, L.; Cras, F. L.; Weill, F. Nat. Mater. 2008, 7 (8), 665. doi: 10.1038/nmat2230
(23) Liu, H.; Strobridge, F. C.; Borkiewicz, O. J.; Wiaderek, K. M.; Chapman, K. W.; Chupas, P. J.; Grey, C. P. Science 2014, 344 (6191), 1252817. doi: 10.1126/science.1252817
(24) Gu, L.; Zhu, C. B.; Li, H.; Yu, Y.; Li, C. L.; Tsukimoto, S.; Maier, J.; Ikuhara, Y. C. J. Am. Chem. Soc. 2011, 133 (13), 4661. doi: 10.1021/ja109412x
(25) Liu, X. S.; Liu, J.; Qiao, R. M.; Yu, Y.; Li, H.; Suo, L. M.; Hu, Y. S.; Chuang, Y. D.; Shu, G. J.; Chou, F. C.; Weng, T. C.; Nordlund, D.; Sokaras, D.; Wang, Y. J.; Lin, H.; Barbiellini, B.; Bansil, A.; Song, X. Y.; Liu, Z.; Yan, S. S.; Liu, G.; Qiao, S.; Richardson, T. J.; Prendergast, D.; Hussain, Z.; Groot, F. M. F. D.; Yang, W. L. J. Am. Chem. Soc. 2012, 134 (33), 13708. doi: 10.1021/ja303225e
(26) Orikasa, Y.; Maeda, T.; Koyama, Y.; Murayama, H.; Fukuda, K.; Tanida, H.; Arai, H.; Matsubara, E.; Uchimoto, Y.; Ogumi, Z. J. Am. Chem. Soc. 2013, 135 (15), 5497. doi: 10.1021/ja312527x
(27) Sun, Y.; Lu, X.; Xiao, R. J.; Li, H.; Huang, X. J. Chem. Mater. 2012, 24 (24), 4693. doi: 10.1021/cm3028324
(28) Xiao, D. D.; Gu, L. Sci. Sin. Chim. 2014, 3 (44), 295. [肖东东,谷 林. 中国科学: 化学, 2014, 3 (44), 295.] doi: 10.1360/032013-269
(29) Cui, Q.; Luo, C. H.; Li, G.; Wang, G. X.; Yan, K. P. Ind. Eng. Chem. Res. 2016, 55 (26), 7069. doi: 10.1021/acs.iecr.6b00023
(30) Churikov, A.; Gribov, A.; Bobyl, A.; Kamzin, A.; Terukov, E. Ionics 2014, 20 (1), 1. doi: 10.1007/s11581-013-0948-4
(31) Ravet, N.; Gauthier, M.; Zaghib, K.; Goodenough, J. B.; Mauger, A.; Gendron, F.; Julien, C. M. Chem. Mater. 2007, 19 (10), 2595. doi: 10.1021/cm070485r
(32) Xiao, Z. W.; Zhang, Y. J.; Hu, G. R. J. Cent. South Univ. 2015, 22 (6), 2043. doi: 10.1007/s11771-015-2727-z
(33) Xiao, Z. W.; Zhang, Y. J.; Hu, G. R. J. Cent. South Univ. 2015, 22 (12), 4507. doi: 10.1007/s11771-015-2999-3
(34) Xiao, Z.; Zhang, Y. J.; Hu, G. R. J. Appl. Electrochem. 2015, 45 (3), 225. doi: 10.1007/s10800-014-0780-1
(35) Weng, S. Y.; Yang, Z. H.; Wang, Q.; Zhang, J.; Zhang, W. X. Ionics 2013, 19 (2), 235. doi: 10.1007/s11581-012-0746-4
(36) Hu, Y. M.; Wang, G. H.; Liu, C. Z.; Chou, S. L.; Zhu, M. Y.; Jin, H. M.; Li, W. X.; Li, Y. Ceram. Int. 2016, 42 (9), 11422. doi: 10.1016/j.ceramint.2016.04.075
(37) Dhindsa, K. S.; Kumar, A.; Nazri, G. A.; Naik, V. M.; Garg, V. K.; Oliveira, A. C.; Vaishnava, P. P.; Zhou, Z. X.; Naik, R. J. Solid State Electrochem. 2016, 20 (8), 2275. doi: 10.1007/s10008-016-3239-y
(38) Reklaitis, J.; Davidonis, R.; Dindune, A.; Valdniece, D.; Jasulaitienė, V.; Baltrūnas, D. Phys. Status Solidi B 2016, 253 (11), 2283. doi: 10.1002/pssb.201600028
(39) Ziolkowska, D. A.; Jasinski, J. B.; Hamankiewicz, B.; Korona, K. P.; Wu, S. H.; Czerwinski. Cryst. Growth Des. 2016, 16 (9), 5006. doi: 10.1021/acs.cgd.6b00575
(40) Xu, C. H.; Wang, L.; He, X. M.; Luo, J.; Shang, Y. M.; Wang, J. L. Int. J. Electrochem. Sci. 2016, 11 (2), 1558
(41) Zhao, H. C.; Song, Y.; Guo, X. D.; Zhong, B. H.; Dong, J.; Liu, H. Acta Phys. -Chim. Sin. 2011, 27 (10), 2347. [赵浩川, 宋 杨,郭孝东, 钟本和, 董 静, 刘 恒. 物理化学学报, 2011, 27 (10), 2347.] doi: 10.3866/PKU.WHXB20110905
(42) Toprakci, O.; Ji, L. W.; Lin, Z.; Toprakci, H. A. K.; Zhang, X. W. J. Power Sources 2011, 196 (18), 7692. doi: 10.1016/j.jpowsour.2011.04.031
(43) Doeff, M. M.; Wilcox, J. D.; Yu, R.; Aumentado, A.; Marcinek, M.; Kostecki, R. J. Solid State Electrochem. 2008, 12 (7), 995. doi: 10.1007/s10008-007-0419-9
(44) Wang, M.; Xue, Y. H.; Zhang, K. L.; Zhang, Y. X. Electrochim. Acta 2011, 56 (11), 4294. doi: 10.1016/j.electacta.2011.01.074
(45) Akiya, N.; Savage, P. E. Chem. Rev. 2002, 102 (8), 2725. doi: 10.1021/cr000668w
(46) Xi, X. L.; Chen, G. L.; Nie, Z. R.; He, S.; Pi, X.; Zhu, X. G.; Zhu, J. J.; Zuo, T. Y. J. Alloy. Compd. 2010, 497 (1), 377. doi: 10.1016/j.jallcom.2010.03.078
(47) Needham, S. A.; Calka, A.; Wang, G.X.; Mosbah, A.; Liu, H, K. Electrochem. Commun. 2006, 8 (3), 434. doi: 10.1016/j.elecom.2005.12.011
(48) Gu, N. Y.; Wang, H.; Li, Y.; Ma, H. Y.; He, X. H.; Yang, Z. Y. J. Solid State Electrochem. 2014, 18 (3), 771. doi: 10.1007/s10008-013-2319-5
(49) Xu, J.; Chen, G.; Xie, C. D.; Li, X.; Zhou, Y. H. Solid State Commun. 2008, 147 (11), 443. doi: 10.1016/j.ssc.2008.07.013
(50) Doan, T. N. L.; Bakenov, Z.; Taniguchi, I. Adv. Powder Technol. 2010, 21 (2), 187. doi: 10.1016/j.apt.2009.10.016
(51) Hwang, B. J.; Hsu, K. F.; Hu, S. K.; Cheng, M. Y.; Chou, T. C.; Tsay, S. Y.; Santhanamd, R. J. Power Sources 2009, 194 (1), 515. doi: 10.1016/j.jpowsour.2009.05.006
(52) Hu, Y. K.; Ren, J. X.; Wei, Q. L.; Guo, X. D.; Tang, Y.; Zhong, B. H.; Liu, H. Acta Phys. -Chim. Sin. 2014, 30 (1), 75. [胡有坤, 任建新, 魏巧玲, 郭孝东, 唐 艳, 钟本和, 刘 恒. 物理化学学报, 2014, 30 (1), 75.] doi: 10.3866/PKU.WHXB201311261
(53) Palomares, V.; Goñi, A.; Muro, I. G. D.; Meatza, I. D.; Bengoechea, Miguel.; Miguel, O.; Rojoa, T. J. Power Sources 2007, 171 (2), 879. doi: 10.1016/j.jpowsour.2007.06.161
(54) Zhu, C.; Yu, Y.; Gu, L.; Weichert, K.; Maier, J. Angew. Chem. Int. Ed. 2011, 50 (28), 6278. doi: 10.1002/anie.201005428
(55) Shao, D. Q.; Wang, J. X.; Dong, X. T.; Yu, W. S.; Liu, G. X.; Zhang, F. F.; Wang, L. M. J. Mater. Sci. -Mater. Electron. 2014, 25 (2), 1040. doi: 10.1007/s10854-013-1684-2
(56) Qiu, Y. J.; Geng, Y. H.; Li, N. N.; Liu, X. L.; Zuo, X. B. Mater. Chem. Phys. 2014, 144 (3), 226. doi: 10.1016/j.matchemphys.2013.12.027
(57) Zhang, C. H.; Liang, Y. Z.; Yao, L.; Qiu, Y. P. J. Alloy. Compd. 2015, 627 (8), 91. doi: 10.1016/j.jallcom.2014.12.067
(58) Patil, K. C.; Aruna, S. T.; Ekambaram, S. Curr. Opin. Solid State Mater. Sci. 1997, 2 (2), 158. doi: 10.1016/S1359-0286 (97)80060-5
(59) Sehrawat, R.; Sil, A. Ionics 2015, 21 (3), 673. doi: 10.1007/s11581-014-1229-6
(60) Mohan, E. H.; Siddhartha, V. Aims Mater. Sci. 2014, 1 (4), 191. doi: 10.3934/matersci.2014.4.191
(61) Vujković, M.; Jugović, D.; Mitrić, M.; Stojkovic, I.; Cvjetićanin, N.; Mentus, Slavko. Electrochim. Acta 2013, 109 (11), 835. doi: 10.1016/j.electacta.2013.07.219
(62) Chu, D. B.; Li, Y.; Song, Q.; Zhou, Y. Acta Phys. -Chim. Sin. 2011, 27 (8), 1863. [褚道葆, 李 艳, 宋 奇, 周 莹. 物理化学学报, 2011, 27 (8), 1863.] doi: 10.3866/PKU.WHXB20110807
(63) Wu, T.; Ma, X.; Liu, X.; Zeng, G.;Xiao, W. Adv. Funct. Mater. 2016, 30 (2), A70. doi: 10.1179/17535557A15Y.000000011
(64) Tang, H.; Xu, J. Mater. Sci. Eng., B 2013, 178 (20), 1503. doi: 10.1016/j.mseb.2013.08.014
(65) Li, Y. C.; Geng, G. G.; Hao, J. H.; Zhang, J. M.; Yang, C. C.; Li, B. J. Electrochim. Acta 2015, 186 (20), 157. doi: 10.1016/j.electacta.2015.10.121
(66) Teja, A. S.; Eckert, C. A. Ind. Eng. Chem. Res. 2000, 39 (12), 4442. doi: 10.1021/ie000915m
(67) Hauthal, W H. Chemosphere 2001, 43 (1), 123. doi: 10.1016/S0045-6535 (00)00332-5
(68) Lee, J.; Teja, A. S. Mater. Lett. 2006, 60 (17), 2105. doi: 10.1016/j.matlet.2005.12.083
(69) Zhang, Y. J.; Yang, Y. F.; Wang, X. Y.; Li, S. S. Chin. J. Chem. Eng. 2014, 22 (2), 234. doi: 10.1016/S1004-9541 (14)60051-3
(70) Rangappa, D.; Sone, K.; Ichihara, M.; Kudo, T.; Honma, I. Chem. Commun. 2010, 46 (40), 7548. doi: 10.1039/c0cc03034a
(71) Xie, M.; Zhang, X. X.; Wang, Y. Z.; Deng, S. X.; Wang, H.; Liu, J. B.; Yan, H.; Laakso, J.; Levänen, E. Electrochim. Acta 2013, 94 (4), 16. doi: 10.1016/j.electacta.2013.01.131
(72) Xie, M.; Zhang, X. X.; Deng, S. X.; Wang, Y. Z.; Wang, H.; Liu, J. B.; Yan, H.; Laakso, J.; Levänen, E. RSC Adv. 2013, 3 (31), 12786. doi: 10.1039/C3RA41133H
(73) Wang, Y. G.; He, P.; Zhou, H. S. Energy Environ. Sci. 2011, 4 (3), 805. doi: 10.1039/c0ee00176g
(74) Zhang, D. Y.; Zhang, P. X.; Lin, M. C.; Liu, K.; Yuan, Q. H.; Xu, Q. M.; Luo, Z. K.; Ren, X. Z. J. Inorg. Mater. 2011, 26 (3), 265. [张冬云, 张培新, 林木崇, 刘 琨, 袁秋华, 许启明, 罗仲宽,任祥忠. 无机材料学报, 2011, 26 (3), 265.] doi: 10.3724/SP.J.1077.2011.00265
(75) Ni, J. F.; Zhou, H, H.; Chen, J. T.; Su, G. Y. Acta Phys. -Chim. Sin. 2004, 20 (6), 582. [倪江锋, 周恒辉, 陈继涛, 苏光耀. 物理化学学报, 2004, 20 (6), 582.] doi: 10.3866/PKU.WHXB20040606
(76) Chen, Y.; Wang, Z. L.; Yu, C. Y.; Xia, D. G.; Wu, Z. Y. Acta Phys. -Chim. Sin. 2008, 24 (8), 1498. [陈 宇, 王忠丽, 于春洋, 夏定国, 吴自玉. 物理化学学报, 2008, 24 (8), 1498.] doi: 10.3866/PKU.WHXB20080829
(77) Mi, C. H.; Cao, G. S.; Zhao, X. B. Chin. J. Inorg. Chem. 2005, 21 (4), 556. [米常焕, 曹高劭, 赵新兵. 无机化学学报, 2005, 21 (4), 556.] doi: 10.3321/j.issn:1001-4861.2005.04.022
(78) Yu, F.; Zhang, J. J.; Yang, Y. F.; Song, G. Z. Chin. J. Inorg. Chem. 2009, 25 (1), 42. [于 锋, 张敬杰, 杨岩峰, 宋广智. 无机化学学报, 2009, 25 (1), 42.] doi: 10.3321/j.issn:1001-4861.2009.01.008
(79) Mi, C. H.; Cao, Y. X.; Zhang, X. G.; Zhao, X. B.; Li, H. L. Powder Technol. 2008, 181 (3), 301. doi: 10.1016/j.powtec.2007.05.017
(80) He, W.; Wei, C. L.; Zhang, X. D.; Wang, Y. Y.; Liu, Q. Z.; Shen, J. X.; Wang, L. Z.; Yue, Y. Z. Electrochim. Acta 2016, 219 (20), 682. doi: 10.1016/j.electacta.2016.10.047
(81) Kobayashi, G.; Nishimura, S. I.; Park, M. S.; Kanno, R.; Yashima, M.; Ida, T.; Yamada, A. Adv. Funct. Mater. 2009, 19 (3), 395. doi: 10.1002/adfm.200801522
(82) Qian, J. F.; Zhou, M.; Cao, Y. L.; Ai, X. P.; Yang, H. X. J. Phys. Chem. C 2010, 114 (8), 3477. doi: 10.1021/jp912102k
(83) Wang, D. Y.; Li, H.; Shi, S. Q.; Huang, X. J.; Chen, L. Q. Electrochim. Acta 2005, 50 (14), 2955. doi: 10.1016/j.electacta.2004.11.045
(84) Hong, J.; Wang, X. L.; Wang, Q.; Omenya, F.; Chernova, N. A.; Whittingham, M. S.; Graetz, J. G. J. Phys. Chem. C 2012, 116 (39), 20787. doi: 10.1021/jp306936t
(85) Zhu, Y. R.; Zhang, R.; Deng, L.; Yi, T. F.; Ye, M. F.; Yao, J. H.; Dai, C. S. Metall. Mater. Trans. E 2015, 2 (1), 33. doi: 10.1007/s40553-014-0041-6
(86) Ma, J.; Li, B. H.; Du, H. D.; Xu, C. J.; Kang, F. Y. J. Solid State Electrochem. 2012, 16 (1), 1. doi: 10.1007/s10008-010-1263-x
(87) Ni, J. F.; Zhou, H. H.; Chen, J. T.; Zhang, X. X. Mater. Lett. 005, 59 (18), 2361. doi: 10.1016/j.matlet.2005.02.080
(88) Lee, S. B.; Cho, S. H.; Heo, J. B.; Aravindan, V.; Kim, H. S.; Lee, Y. S. J. Alloy. Compd. 2009, 488 (1), 380. doi: 10.1016/j.jallcom.2009.08.144
(89) Li, Y. C.; Hao, J. H.; Geng, G. W.; Wang, Y. F.; Shang, X. K.; Yang, C. C.; Li, B. J. RSC Adv. 2015, 5 (84), 68681. doi: 10.1039/C5RA11680E
(90) Yuan, H.; Wang, X. Y.; Wu, Q.; Shu, H. B.; Yang, X. K. J. Alloy. Compd. 2016, 675 (5), 187. doi: 10.1016/j.jallcom.2016.03.065
(91) Zhao, N. N.; Li, Y. S.; Zhi, X. K.; Wang, L.; Zhao, X. X.; Wang, Y. M.; Liang, G. C. J. Rare Earths 2016, 34 (2), 174. doi: 10.1016/S1002-0721(16)60011-X
(92) Naik, A.; Zhou, J.; Gao, C.; Liu, G. Z.; Wang, L. Mater. Sci.-Poland 2016, 33 (4), 742. doi: 10.1515/msp-2015-0087
(93) Wang, Y. R.; Yang, Y. F.; Hu, X.; Yang, Y. B.; Shao, H. X. J. Alloy. Compd. 2009, 481 (1), 590. doi: 10.1016/j.jallcom.2009.03.033
(94) Johnson, I. D.; Lübke, M.; Wu, O. Y.; Makwana, N. M.; Smales, G. J.; Islam, H. U.; Dedigama, R. Y.; Gruar, R. I.; Tighe, C. J.; Scanlon, D. O.; Corà, F.; Brett, D. J. L.; Shearing, P. R.; Darr, J. A. J. Power Sources 2016, 302 (20), 410. doi: 10.1016/j.jpowsour.2015.10.068
(95) Li, L. J.; Li, X. H.; Wang, Z. X.; Wu, L.; Zheng, J. C.; Guo, H. J. J. Phys. Chem. Solids 2009, 70 (1), 238. doi: 10.1016/j.jpcs.2008.10.012
(96) Naik, A.; Zhou, J.; Gao, C.; Liu, G. Z.; Wang, L. J. Energy Inst. 2016, 89 (1), 21. doi: 10.1016/j.joei.2015.01.013
(97) Johnson, I. D.; Blagovidova, E.; Dingwall, P. A.; Brett, D. J. L.; Shearing, P. R.; Darr, J. A. J. Power Sources 2016, 326 (15), 476. doi: 10.1016/j.jpowsour.2016.06.128
(98) Yang, S. T.; Liu, Y. X.; Yin, Y. H.; Wang, H.; Wang, T. Chin. J. Inorg. Chem. 2007, 23 (7), 1165. [杨书廷, 刘玉霞, 尹艳红,王 辉, 王 涛. 无机化学学报, 2007, 23 (7), 1165.] doi: 10.3321/j.issn:1001-4861.2007.07.007
(99) Naik, A.; Ponnappa, S. C. Dalton Trans. 2016, 45 (19), 8021. doi: 10.1039/C6DT00331A
(100) Shu, H. B.; Wang, X. Y.; Wen, W. C.; Liang, Q. Q.; Yang, X. K.; Wei, Q. L.; Hu, B. A.; Liu, L.; Liu, X.; Song, Y. F.; Zho, M.; Bai, Y. S.; Jiang, L. L.; Chen, M. F.; Yang, S. Y.; Tan, J. L.; Liao, Y. Q.; Jiang, H. M. Electrochim. Acta 2013, 89 (1), 479. doi: 10.1016/j.electacta.2012.11.081
(101) Xu, Y.; Zhao, M. H.; Sun, B. Solid State Ionics 2016, 291, 14. doi: 10.1016/j.ssi.2016.04.008
(102) Chung, S. Y.; Bloking, J. T.; Chiang, Y. M. Nat. Mater. 2002, 1 (2), 123. doi: 10.1038/nmat732
(103) Liao, X. Z.; He, Y. S.; Ma, Z. F.; Zhang, X. M.; Wang, L. J. Power Sources 2007, 174 (2), 720. doi: 10.1016/j.jpowsour.2007.06.146
(104) Sun, C. S.; Zhang, Y.; Zhang, X. J.; Zhou, Z. J. Power Sources 2010, 195 (11), 3680. doi: 10.1016/j.jpowsour.2009.12.074
(105) Li, H.; Wang, Z. X.; Chen, L. Q.; Huang, X. J. Adv. Mater. 2009, 21 (45), 4593. doi: 10.1002/adma.200901710
(106) Abbate, M.; Lala, S. M.; Montoro, L. A.; Rosolenb, J. M. Electrochem. Solid-State Lett. 2005, 8 (6), A288. doi: 10.1149/1.1895286
(107) Lu, J. B.; Tang, Z. L.; Zhang, Z. T.; Jin, Y. Z. Acta Phys. -Chim. Sin. 2005, 21 (3), 319. [卢俊彪, 唐子龙, 张中太, 金永拄. 物理化学学报, 2005, 21 (3), 319.] doi: 10.3866/PKU.WHXB20050319
(108) Yu, W.; Ou, G.; Qi, L. H.; Wu, H. J. Am. Ceram. Soc. 2016, 99 (10), 3214. doi: 10.1111/jace.14426
(109) Zaghib, K.; Mauger, A.; Goodenough, J. B.; Gendron, F.; Julien, C. M. Chem. Mater. 2007, 19 (15), 3740. doi: 10.1021/cm0710296
(110) Harrison, K. L.; Bridges, C. A.; Paranthaman, M. P.; Segre, C. U.; Katsoudas, J.; Maroni, V. A.; Idrobo, J. C.; Goodenough, J. B.; Manthiram, A. Chem. Mater. 2013, 25 (5), 768. doi: 10.1021/cm303932m
(111) Herle, P. S.; Ellis, B.; Coombs, N.; Nazar, L. F. Nat. Mater. 2004, 3 (3), 147. doi: 10.1038/nmat1063
(112) Delacourt, C.; Wurm, C.; Laffont, L.; Leriche, J. B.; Masquelier, C. Solid State Ionics 2006, 177, 333. doi: 10.1016/j.ssi.2005.11.003
(113) Park, K. S.; Xiao, P. H.; Kim, S. Y.; Dylla, A.; Choi, Y. M.; Henkelman, G.; Stevenson, K. J.; Goodenough, J. B. Chem. Mater. 2012, 24 (16), 3212. doi: 10.1021/cm301569m
(114) Ravet, N.; Goodenough, J. B.; Besner, S.; Simoneau, M.; Hovington, P.; Armand, M. J. Electrochem. Soc. Abstr. 1999, 99-2, 172.
(115) Sun, L. N.; Deng, Q. W.; Fang, B.; Li, Y. L.; Deng, L. B.; Yang, B.; Ren, X. Z.; Zhang, P. X. CrystEngComm 2016, 18 (39), 7537. doi: 10.1039/C6CE01681B
(116) Wang, K.; Cai, R.; Yuan, T.; Yu, X.; Ran, R.; Shao, Z. P. Electrochim. Acta 2009, 54 (10), 2861. doi: 10.1016/j.electacta.2008.11.012
(117) Liu, X. H.; Zhao, Z. W. Powder Technol. 2010, 197 (3), 309. doi: 10.1016/j.powtec.2009.09.019
(118) Shin, H. C.; Cho, W. I.; Jang, H. J. Power Sources 2006, 159 (2), 1383. doi: 10.1016/j.jpowsour.2005.12.043
(119) Sun, C.; Yan, L. M.; Yue, B. H. Acta Phys. -Chim. Sin. 2013, 29 (8), 1666. [孙 超, 严六明, 岳宝华. 物理化学学报, 2013, 29 (8), 1666.] doi: 10.3866/PKU.WHXB201304232
(120) Ni, J. F.; Morishita, M.; Kawabe, Y.; Watada, M.; Takeichi, N.; Sakai, T. J. Power Sources 2010, 195 (9), 2877. doi: 10.1016/j.jpowsour.2009.11.017
(121) Gaberscek, M.; Dominko, R.; Bele, M.; Remskar, M.; Hanzel, D.; Jamnik, J. Solid State Ionics 2005, 176, 1801. doi: 10.1016/j.ssi.2005.04.034
(122) Zhang, H. Y.; Chen, Y. T.; Zheng, C. C.; Zhang, D. F.; He, C. H. Ionics 2015, 21 (7), 1813. doi: 10.1007/s11581-014-1354-2
(123) Yu, H. M.; Zheng, W.; Cao, G. S.; Zhao, X. B. Acta Phys. -Chim. Sin. 2009, 25 (11), 2186. [余红明, 郑 威, 曹高劭, 赵新兵.物理化学学报, 2009, 25 (11), 2186.] doi: 10.3866/PKU.WHXB20091113
(124) Kuwahara, A.; Suzuki, S.; Miyayama, M. Ceram. Int. 2008, 34 (4), 863. doi: 10.1016/j.ceramint.2007.09.037
(125) Du, Y. H.; Tang, Y. F.; Huang, F. Q.; Chang, C. k. RSC Adv. 2016, 6 (57), 52279. doi: 10.1039/C6RA08937B
(126) Li, Q. R.; Zhou, Z. F.; Liu, S. S.; Zhang, X. X. Ionics 2016, 22 (7), 1027. doi: 10.1007/s11581-016-1636-y
(127) Lu, J. B.; Le, B.; Tang, Z. L.; Zhang, Z. T.; Li, J. R.; Shen, W. C. Rare Metal Mat. Eng. 2005, 34 (z2), 673. [卢俊彪, 乐 斌, 唐子龙, 张中太, 李俊荣, 沈万慈. 稀有金属材料与工程, 2005, 34 (z2), 673.] doi: 10.3321/j.issn:1002-185X.2005.z2.016
(128) Yang, W. Y.; Zou, M. Z.; Zhao, G. Y.; Hong, Z. S.; Feng, Q.; Li, J. X.; Lin, Y. B.; Huang, Z. G. Solid State Ionics 2016, 292, 103. doi: 10.1016/j.ssi.2016.05.017
(129) Saliman, M. A.; Okawa, H.; Takai, M.; Ono, Y.; Kato, T.; Sugawara, K.; Sato, M. Jpn. J. Appl. Phys. 2016, 55 (7S1), 07KE05. doi: 10.7567/JJAP.55.07KE05
(130) Zhao, S. X.; Li, Y. D.; Ding, H.; Li, B. H.; Nan, C. W. J. Inorg. Mater. 2013, 28 (11), 1265. [赵世玺, 李颖达, 丁 浩, 李宝华,南策文. 无机材料学报, 2013, 28 (11), 1265.] doi: 10.3724/SP.J.1077.2013.13136
(131) Xu, Y. P.; Mao, J. J. Mater. Sci. 2016, 51 (22), 10026. doi: 10.1007/s10853-016-0229-5
(132) Tang, H.; Tan, L.; Jun, X. U. T. Nonferr. Metal. Soc. 2013, 23 (2), 451. doi: 10.1016/S1003-6326 (13)62484-X
(133) Zhang, X. P.; Guo, H. J.; Li, X. H.; Wang, Z. X.; Peng, W. J.; Wu, L. Chem. J. Chin. Univ. 2012, 33 (2), 236. [张晓萍, 郭华军, 李新海, 王志兴, 彭文杰, 伍 凌. 高等学校化学学报, 2012, 33 (2), 236.] doi: 10.3969/j.issn.0251-0790.2012.02.005 (134) Ma, Z. P.; Shao, G. J.; Wang, X.; Song, J. J.; Wang, G. L. Ionics 2013, 19 (12), 1861. doi: 10.1007/s11581-013-0950-x
(135) Ma, Z. P.; Shao, G. J.; Qin, X. J.; Fan, Y. Q.; Wang, G. L.; Song, J. J.; Liu, T. T. J. Power Sources 2014, 269 (4), 194. doi: 10.1016/j.jpowsour.2014.06.157
(136) Ma, Z. P.; Peng, Y. S.; Wang, G. L.; Fan, Y. Q.; Song, J. J.; Liu, T. T.; Qin, X. J.; Shao, G. J. Electrochim. Acta 2015, 156 (12), 77. doi: 10.1016/j.electacta.2015.01.015
(137) Bai, Y. M.; Qiu, P.; Wen, Z. L.; Han, S. C. J. Alloy. Compd. 2010, 508 (1), 1. doi: 10.1016/j.jallcom.2010.05.173
(138) Huang, Y. H.; Goodenough, J. B. Chem. Mater. 2008, 20 (23), 7237. doi: 10.1021/cm8012304
(139) Sehrawat, R.; Sil, A. J. Mater. Sci.: Mater. Electron. 2015, 26 (7), 5175. doi: 10.1007/s10854-015-3048-6
(140) Yamada, A.; Chung, S. C.; Hinokuma, K. J. Electrochem. Soc. 2001, 148 (3), A224. doi: 10.1149/1.1348257
(141) Wu, J.; Dathar, G. K. P.; Sun, C. W.; Theivanayagam, M. G.; Applestone, D.; Dylla, A. G.; Manthiram, A.; Henkelman, G.; Goodenough, J. B.; Stevenson, K. J. Nanotechnology 2013, 24 (42), 424009. doi: 10.1088/0957-4484/24/42/424009
(142) Huang, H.; Yin, S. C.; Nazar, L. F. Electrochem. Solid-State Lett. 2001, 4 (10), A170. doi: 10.1149/1.1396695
(143) Saravanan, K.; Balaya, P.; Reddy, M. V.; Chowdari, B. V. R.; Vittal, J. J. Energy Environ. Sci. 2010, 3 (4), 457. doi: 10.1039/B923576K
(144) Hong, S. A.; Kim, S. J.; Chung, K. Y.; Chun, M. S.; Lee, B. G.; Kim, J. J. Supercrit. Fluids 2013, 73, 70. doi: 10.1016/j.supflu.2012.11.008
(145) Liu, T. F.; Zhao, L.; Wang, D. L.; Zhu, J. S.; Wang, B.; Guo, C. F. RSC Adv. 2014, 4 (20), 10067. doi: 10.1039/C3RA46975A
(146) Liu, Z. X.; Xu, B.; Xing, Y.; Li, J. J.; Zhang, L. S.; Wang, L. Z.; Fang, S. M. J. Nanopart. Res. 2015, 17 (3), 163. doi: 10.1007/s11051-015-2969-6
(147) Viji, M.; Swain, P.; Mocherla, P. S. V.; Sudakar, C. RSC Adv. 2016, 6 (46), 39710. doi: 10.1039/C6RA04468A
(148) Wilcox, J. D.; Doeff, M. M.; Marcinek, M.; Kostecki, R. J. Electrochem. Soc. 2007, 154 (5), A389. doi: 10.1149/1.2667591
(149) Yu, F.; Zhang, L. L.; Li, Y. C.; An, Y. X.; Zhu, M. Y.; Dai, B. RSC Adv. 2014, 4 (97), 54576. doi: 10.1039/C4RA10899J
New Research Progress of the Electrochemical Reaction Mechanism, Preparation and Modification for LiFePO4
ZHANG Ying-Jie ZHU Zi-Yi DONG Peng QIU Zhen-Ping LIANG Hui-Xin LI Xue*
(National and Local Joint Engineering Laboratory for Lithium-ion Batteries and Materials Preparation Technology, Key Laboratory of Advanced Battery Materials of Yunnan Province, Kunming University of Science and Technology, Kunming 650000, P. R. China)
Lithium-ion batteries have been extensively studied due to their excellent electrochemical performance as an effective energy storage device for sustainable energy sources. The key to the development and application of this technology is the improvement of electrode materials. LiFePO4has captured the attention of researchers both home and abroad as a potential cathode material for lithium-ion batteries because of its long cycle life, energy density, stable charge/discharge performance, good thermal stability, high safety, light weight and low toxicity. However, there are still some technical bottlenecks in the application of LiFePO4, such as relatively low conductivity, low diffusion coefficient of lithium ions, and low tap density. Moreover, the cycle performance, low-temperature characteristics, and rate performance are not ideal, restricting its application and development. In recent years, researchers have sought to solve these problems by improving the preparation process and attempting related modifications. In this paper, we have provided a systemic review of the structure, electrochemical reaction mechanism, preparation, and modification of LiFePO4. The main problems associated with LiFePO4cathode materials and possible solutions are discussed. We have also investigated the future research direction and application prospect of LiFePO4cathode materials.
December 21, 2016; Revised: March 13, 2017; Published online: April 11, 2017.
O646
10.3866/PKU.WHXB201704114
*Corresponding authors. Email: 438616074@qq.com; Tel: +86-18808804528.
The project was supposed by the National Natural Science Foundation of China (51604132).
国家自然科学基金(51604132)资助项目
© Editorial office of Acta Physico-Chimica Sinica
