活性MgO的制备及其去除溶液中Th(Ⅳ)的性能
2017-06-19秦启凤李小燕刘晴晴邹宇轩张卫民
秦启凤,李小燕,刘晴晴,邹宇轩,杨 波,张卫民,李 寻
(东华理工大学 省部共建核资源与环境国家重点实验室培育基地,江西 南昌 330013)
活性MgO的制备及其去除溶液中Th(Ⅳ)的性能
秦启凤,李小燕,刘晴晴,邹宇轩,杨 波,张卫民,李 寻
(东华理工大学 省部共建核资源与环境国家重点实验室培育基地,江西 南昌 330013)
研究了以微波为热源,采用均匀沉淀法制备活性MgO,并用所制备的MgO去除溶液中的Th(Ⅳ),考察了溶液pH、反应温度、接触时间、固液质量体积比、溶液中的Th钍初始质量浓度对Th(Ⅳ)去除效果的影响。结果表明:在pH=2.5、温度25 ℃、接触时间60 min、固液质量体积比0.15 g/L、Th(Ⅳ)初始质量浓度50 mg/L条件下,活性MgO对溶液中Th(Ⅳ)去除率达98.91%,去除效果较好;用少量活性MgO处理酸性含钍放射性废水即可取得很好的效果,经济环保、操作简单,具有较好的应用前景;推测活性MgO去除溶液中Th(Ⅳ)的机制为以化学沉淀为主,表面物理吸附为辅。
活性MgO;制备;钍;去除
钍可用于核能发电,弥补铀资源的不足[1-3]。钍常与稀土和铀伴生[3-4],在开发稀土矿和铀矿过程中,钍被释放出来。钍工业回收率较低,铀水冶过程中75%的钍留在尾矿中[4-5],形成含钍放射性废水废渣。矿山排放的废水中一般含有大量硫酸根(pH在2~4之间),酸性很强[6],酸性含钍放射性废水随地下水进入生物圈会使土壤酸化,威胁水生生物,对周围环境造成放射性污染等[6-7]。
目前,酸性废水的处理一般采用中和沉淀法,加入氢氧化钠、石灰石等碱性物质中和酸,使生成氢氧化物沉淀,该法操作简单,处理费用低[8]。为了避免二次污染,对碱性物质的选择尤为重要。MgO是一种绿色氧化物,对环境无害,水合后表面产生大量OH-而呈碱性[9],生成的Mg(OH)2是一种绿色安全的中和剂,具有缓冲性[10]。活性MgO表面有大量缺陷,这些缺陷使其具有较强的吸附性能和活性[11-12]。试验模拟酸性含钍放射性废水,利用微波辅助技术制备活性MgO,并研究了其在不同条件下从溶液中去除Th(Ⅳ)的性能,以期为酸性含钍放射性废水的综合治理提供可供选择的物质。
1 试验材料与方法
1.1 试剂与仪器
主要试剂:氯化镁(MgCl2·6H2O)、十二烷基硫酸钠、尿素、硝酸钍(Th(NO3)4·4H2O)、无水乙醇、草酸、盐酸、偶氮胂Ⅲ,均为分析纯,国药集团化学试剂有限公司产品。
主要仪器:MSP-6600型微波消解仪,北京瑞利分析仪器有限公司;TDL-40B型离心机,上海安亭仪器厂;pHS-3C型pH计,上海智光仪器仪表有限公司;CP124C型电子天平,奥豪斯仪器(上海)有限公司;DZF-6020型真空干燥箱,上海三发科学仪器有限公司;SHZ-82A型气浴恒温振荡箱,江苏省金坛市荣华仪器有限公司;2MF-6型一体化智能马弗炉,常州市方嘉电子仪器有限公司;722型分光光度计,浙江托普仪器有限公司。
钍标准溶液:准确称量(Th(NO3)4·4H2O) 2.379 3 g于坩埚内,加入少量蒸馏水溶解,再加入10 mL盐酸,在电炉上加热溶解蒸发至近干。冷却后,加入20 mL 0.1 mol/L盐酸溶液溶解并移入1 000 mL容量瓶中,用蒸馏水定容、摇匀,配得质量浓度为1 000 mg/L的钍标准溶液。此溶液稀释后用于后续试验。
1.2 活性MgO的制备
活性MgO的传统制备方法是水热合成法。微波加热法作为一种新型方法,相比传统水热法具有加热速度快、效率高的优点。在微波消解仪中加热80 min即能制得纯度高、粒径较小的前驱物[13],大大缩短制备样品所需时间。
借助智能型微波消解仪三段式提供热源,采用均匀沉淀法制备活性MgO。以氯化镁为原料,尿素为沉淀剂,在高温高压条件下,尿素水解生成构晶离子OH-,在二氧化碳和水存在条件下,OH-与Mg2+结合生成碱式碳酸镁晶核[14]。加入少量表面活性剂(十二烷基硫酸钠)包覆在其表面,可以控制颗粒尺寸防止颗粒团聚,从而使颗粒缓慢成长,获得纯度较高的碱式碳酸镁Mg5(CO3)4(OH)2·4H2O[15-16]。碱式碳酸镁在400 ℃下煅烧1 h即得活性MgO。制备过程中的反应如下:
2OH-+CO2↑;
(1)
Mg5(CO3)4(OH)2·4H2O;
(2)
H2O+CO2↑。
(3)
1.3 试验过程
准确称取一定量活性MgO于250 mL锥形瓶中,加入一定质量浓度的Th(Ⅳ)溶液,用NaOH或HNO3溶液调节溶液pH,在不同温度下放入恒温振荡箱中振荡一段时间后取出,置于4 000 r/min离心机中离心5 min后,取上清液用偶氮胂Ⅲ分光光度法在664 nm处测其分光度,根据标准曲线求出Th(Ⅳ)质量浓度,按式(4)、(5)分别计算活性MgO对Th(Ⅳ)的去除率(R)和吸附容量(Q)。

(4)

(5)
式中:ρ0,ρe分别为溶液中钍的初始质量浓度和反应后质量浓度,mg/L;m为活性MgO质量,g;V为溶液体积,L。
2 试验结果与讨论
2.1 活性MgO的表征
2.1.1 扫描电镜(SEM)表征
反应前、后活性MgO的SEM照片如图1所示。可以看出:所制备活性MgO呈片状,其表面有很多蓬松的片状物堆积,空隙较大,整体呈花瓣状,比表面积较大;反应后,活性MgO表面变得比较密实,之前蓬松的片状沉积物聚集起来,空隙变小,发生了团聚,一定程度上说明其发生了物理吸附。

图1 活性MgO的SEM照片
2.1.2 能谱(EDS)表征
图2为活性MgO的EDS表征结果,活性MgO吸附Th(Ⅳ)前、后的元素的EDS分析结果见表1。可以看出:反应后的MgO中有Th元素,Th质量分数由反应前的0增加到6.63%,原子质量分数比由0增加到0.51%,表明活性MgO对Th(Ⅳ)发生了吸附作用。
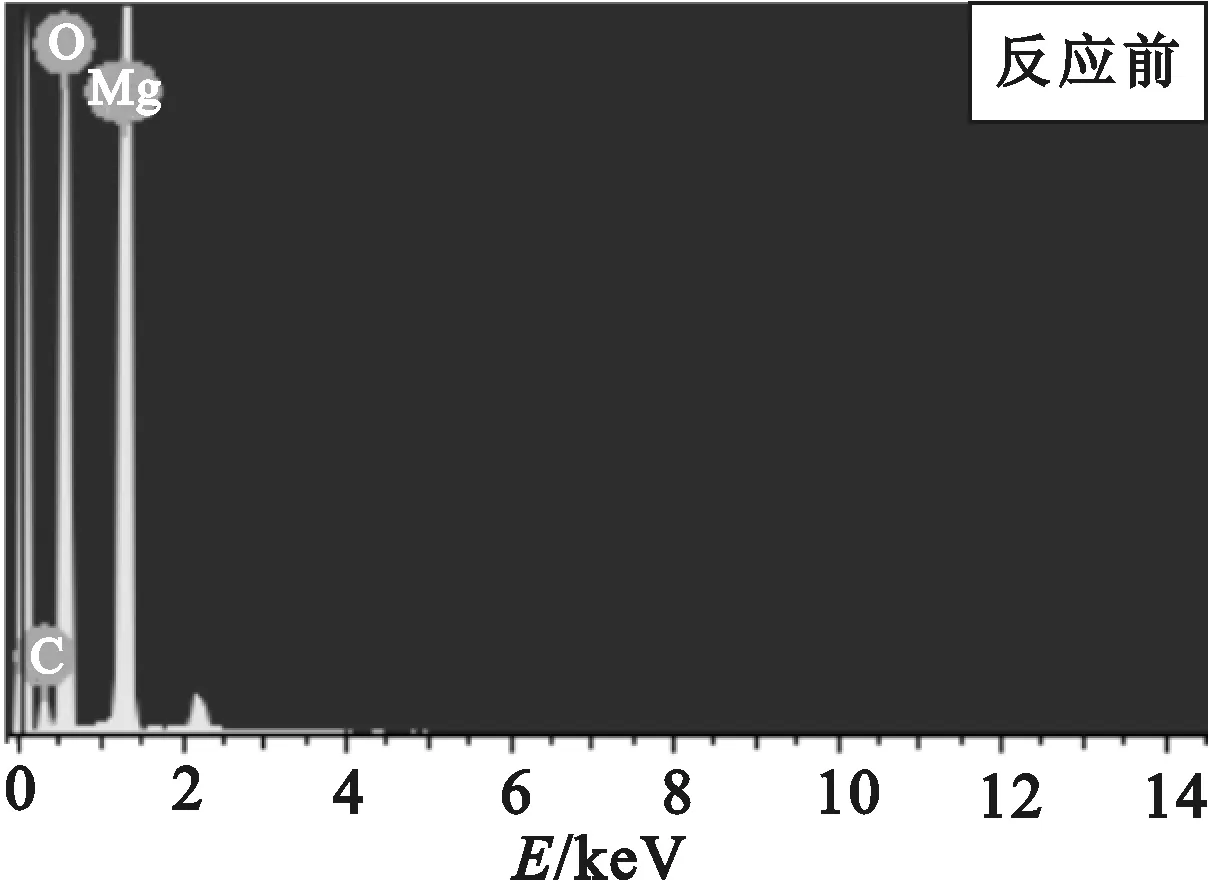

图2 活性MgO的EDS分析曲线

表1 活性MgO吸附Th(Ⅳ)前、后元素的EDS分析结果 %
2.1.3 X射线衍射(XRD)分析
图3为活性MgO的XRD分析结果。
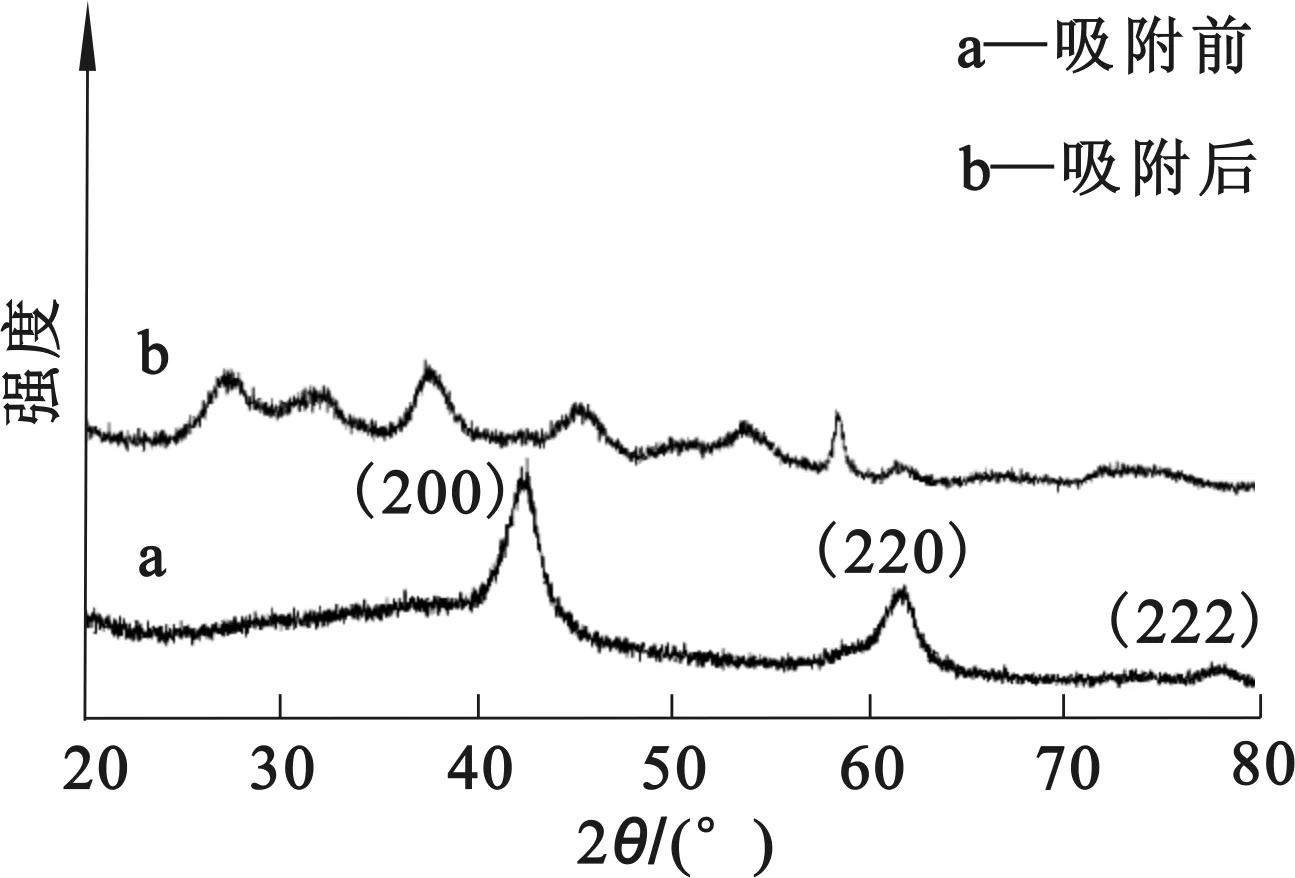
图3 吸附Th(Ⅳ)前后活性MgO的XRD图谱
由图3看出:所制备的活性MgO在2θ=42.49°、62.01°、78.39°条件下的衍射峰与MgO(JCPDS 74-1255)标准卡的衍射峰峰面(200)、(220)、(222)吻合,说明所制备的活性MgO纯度较高;吸附Th(Ⅳ)后的活性MgO的衍射峰发生位移,且出现了几个新峰,说明其晶体结构发生了改变,MDI Jade物相分析反应后新生成的物质主要是Th(OH)4,即吸附过程中发生了化学反应。
2.2 活性MgO去除溶液中的Th(Ⅳ)
2.2.1 溶液pH对活性MgO去除Th(Ⅳ)的影响
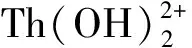
在25 ℃条件下,向装有50 mL钍溶液的锥形瓶中加入7.5 mg活性MgO,溶液中Th(Ⅳ)初始质量浓度为50 mg/L,调节溶液pH,在恒温振荡器中振荡60 min,后续步骤按1.3操作。溶液pH对溶液中Th(Ⅳ)去除率的影响试验结果见表2。

表2 溶液pH对Th(Ⅳ)去除率的影响
由表2看出,体系中加入少量活性MgO,溶液pH发生很大变化:pH=2时,酸性过强,破坏了MgO的表面结构,将其直接溶解,所以钍去除率很低;pH≥2.5后,反应后的溶液都呈碱性,钍去除率达94%以上。由于活性MgO比表面积较大,表面Mg2+较多,与水反应后生成Mg(OH)2,溶液中OH-浓度增大,溶液pH增大,Th(Ⅳ)在碱性环境中水解沉淀为Th(OH)4,易从溶液中沉淀去除;初始pH=2.5时,加入活性MgO后,溶液pH升至9.26,钍去除率达95.22%,而且在去除Th(Ⅳ)的同时改善了酸性水质。利用活性MgO水合后可大幅提高溶液pH的特点处理酸性含钍废水,大部分钍形成沉淀,少部分钍在MgO表面发生物理吸附。后续试验中控制溶液pH=2.5。
2.2.2 固液质量体积比对活性MgO去除Th(Ⅳ)的影响
在25 ℃条件下,调节溶液pH=2.5,Th(Ⅳ)初始质量浓度为50 mg/L,溶液体积50 mL,接触时间60 min,固液质量体积比对Th(Ⅳ)去除率的影响试验结果如图4所示。可以看出:Th(Ⅳ)去除率随固液质量体积比增大快速提高,在0.15 g/L条件下,即活性MgO用量为7.5 mg时,Th(Ⅳ)去除率达最大98.91%;继续增大氧化镁用量,Th(Ⅳ)去除率保持较高水平不再变化。综合考虑,确定活性MgO适宜用量为7.5 mg/50 mL。
2.2.3 接触时间对活性MgO去除Th(Ⅳ)的影响
溶液体积50 mL,Th(Ⅳ)初始质量浓度50 mg/L,温度25 ℃,活性MgO用量7.5 mg,溶液pH=2.5,接触时间对Th(Ⅳ)去除率的影响试验结果如图5所示。

图5 接触时间对Th(Ⅳ)去除率的影响
由图5看出:Th(Ⅳ)去除率随接触时间延长快速升高,在接触60 min时达最高98.06%;继续延长接触时间,钍去除率变化不大。这主要是因为随反应进行,活性MgO表面不断产生OH-,产生的OH-首先要中和溶液中的H+,待溶液pH升高后才沉淀大部分Th(Ⅳ),所以接触时间小于30 min时,Th(Ⅳ)去除率很低,30 min后,Th(Ⅳ)去除率迅速升高,接触60 min时反应达到平衡。
2.2.4 Th(Ⅳ)初始质量浓度对去除Th(Ⅳ)的影响
溶液体积50 mL,溶液pH=2.5,活性MgO用量7.5 mg,接触时间60 min,Th(Ⅳ)初始质量浓度对Th(Ⅳ)去除率的影响试验结果如图6所示。

图6 Th(Ⅳ)初始质量浓度对Th(Ⅳ)去除率的影响
由图6看出:Th(Ⅳ)初始质量浓度<200 mg/L时,活性MgO对Th(Ⅳ)去除效果较好,Th(Ⅳ)去除率在97%以上。活性MgO表面积很大,吸附位点多,活性很强,能将Th(Ⅳ)从初始质量浓度较高的溶液中载带下来。吸附容量(Q)随Th(Ⅳ)初始质量浓度升高而迅速增大,在Th(Ⅳ)初始质量浓度为500 mg/L时,Q达最大169.10 mg/g;Th(Ⅳ)初始质量浓度达1 000 mg/L时,Q降至59.80 mg/g,说明吸附位点逐渐被占满,吸附达到饱和。实际生产过程中,Th(Ⅳ)质量浓度不会富集至很高(超过200 mg/L),所以,在含Th(Ⅳ)废水中加入少量活性MgO就可将Th(Ⅳ)从溶液中去除,使废水达到排放标准。试验中,模拟废水中Th(Ⅳ)质量浓度为50 mg/L。
2.2.5 温度对活性MgO去除Th(Ⅳ)的影响
溶液中Th(Ⅳ)初始质量浓度为50 mg/L,pH=2.5,活性MgO用量7.5 mg,接触时间60 min,温度对Th(Ⅳ)去除率的影响试验结果如图7所示。

图7 温度对Th(Ⅳ)去除率的影响
由图7看出:温度在20~40 ℃范围内,活性MgO对Th(Ⅳ)的去除效果较好;温度升至40 ℃后,Th(Ⅳ)去除率迅速下降。大多数情况下,沉淀物的溶解过程吸热,当温度升高时溶解度升高[21],使溶液中已经沉淀的物质部分溶解,导致Th(Ⅳ)去除率下降,所以温度不宜过高。因此,从节能角度考虑,选择温室即可。
3 结论
利用微波加热均匀沉淀法制备活性MgO是可行的,所得MgO表面呈片状堆积,比表面积较大,对溶液中的Th(Ⅳ)具有较强的吸附作用。在室温下,控制溶液pH=2.5、接触时间60 min、固液质量体积比0.15 g/L、Th(Ⅳ)初始质量浓度50 mg/L,活性MgO对溶液中Th(Ⅳ)的去除率可高达98.91%。活性MgO水合后能使酸性溶液pH大幅上升,在处理矿山酸性废水中可以充分利用这个优势,用少量活性MgO就能达到很好的效果,经济环保。
活性MgO对溶液中Th(Ⅳ)的去除机制推测为大部分为化学沉淀,少部分为表面物理吸附。
[1] 张书成,刘平,仉宝聚.钍资源及其利用[J].世界核地质科学,2005,22(2):98-103.
[2] 王攀峰,聂文斌,花榕,等.骨粉对放射性废水中钍的吸附行为研究[J].原子能科学技术,2015,49(7):19-23.
[3] 刘建亮.一维钛基活性材料的合成、表征及对Th(Ⅳ)的吸附研究[D].抚州:东华理工大学,2013.
[4] 吴婉滢,姚广超,张晓文,等.改性稻草对钍的吸附行为[J].核技术,2015,38(4):1-7.
[5] 周敏.钍在铀水冶过程中的迁移行为研究[D].抚州:东华理工大学,2013.
[6] 潘科,李正山.矿山酸性废水治理技术及其发展趋势[J].四川环境,2007,26(5):83-84.
[7] PEPPAS A,KOMNITSAS K,HALIKIA I.Use of organic covers for acid mine drainage control [J].Minerals Engineering,2000,13(5):563-574.
[8] 赵永斌.微电解中和沉淀法处理酸性矿区地下水的研究[D].广州:广东工业大学,2002.
[9] 鲁俊文,王维,张爱清,等.活性氧化镁的吸附及分解性能研究进展[J].硅酸盐通报,2011,30(5):112-116.
[10] 王路明.氢氧化镁对酸性废水处理的研究[J].盐业与化工,2008,37(6):21-25.
[11] XU Y J,ZHANG Y F,LU N X,et al.Cluster models study of CH2O adsorption and dissociation at defect sites of MgO(001) surface[J].Phys B Condens Matter,2004,348(1/2/3/4):190-197.
[12] 李奕,李俊篯,吴立明,等.MgO缺陷和不规则表面吸附CO的能带和电子结构研究[J].化学学报,2000,58(8):975-980.
[13] 宋羽,王永钱,袁曦明.微波在活性稀土发光材料制备中的应用[J].化工新型材料,2006,34(3):12-14.
[14] 柴多里,张柠,杨保俊.花瓣状活性氢氧化镁及氧化镁的微波辅助合成[J].无机盐工业,2011,43(5):40-43.
[15] 袁培.活性氧化镁化学合成与吸附性能研究[D].长沙:中南大学,2014.
[16] 胡章文,汪兵,单承湘,等.活性氢氧化镁的制备[J].盐湖研究,2005,13(4):47-50.
[17] 盛国栋,杨世通,郭志强,等.活性材料和活性技术在核废料处理中的应用研究进展[J].核化学与放射化学,2012,34(6):321-330.
[18] 许君政,范桥辉,白洪彬,等.离子强度、温度、pH和腐殖酸浓度对Th(Ⅳ)在凹凸棒石上吸附的影响[J].核化学与放射化学,2009,31(3):179-185.
[19] ZENG Yunhang,LIAO Xuepin,HE Qiang,et al.Recovery of Th(Ⅳ) from aqueous solution by reassembled collagen-tannin fiber adsorbent[J].Journal of Radioanalytical and Nuclear Chemistry,2009,280(1):91-98.
[20] KAYNARA Ü H,AYVACΙKLΙ M,HÇSÖNMEZ Ü,et al.Removal of thorium(Ⅳ) ions from aqueous solution by a novel nanoporons ZnO:isotherms,kinetic and thermodynamic studies[J].Journal of Environmental Radioactivity,2015,150:145-151.
[21] 王祥云,刘远方.核化学与放射化学[M].北京:北京大学出版社,2007.
Preparation of Active Magnesium Oxide by Microwave Heating and Removal of Th(Ⅳ) in Aqueous Solution
QIN Qifeng,LI Xiaoyan,LIU Qingqing,ZOU Yuxuan,YANG Bo,ZHANG Weiming,LI Xun
(StateKeyLaboratoryBreedingBaseofNuclearResourcesandEnvironment,EastChinaUniversityofTechnology,Nanchang330013,China)
Preparation of active magnesium oxide by microwave heating and homogeneous precipitation method was investigated,then remove Th(Ⅳ) from aqueous solution using the active MgO was also investigated.The effects of solution pH,temperature,contact time,solid-to-liquid ratio and initial concentrations of Th(Ⅳ) on removal of Th(Ⅳ) were examined,and the properties of active MgO were characterized.The result show that active MgO has very good removal efficiency for Th(Ⅳ) under the conditions of pH=2.5,60 min of contact time,0.15 g/L of solid-to-liquid ratio,50 mg/L of initial concentration of Th(Ⅳ),the highest removal rate reaches 98.91%.Using a small amount of the active MgO to treat acidic radioactive wastewater containing thorium can achieve very good effects with economic and environmental protection,simple operation.The mechanism of removal of Th(Ⅳ) by the active MgO is presumed to be mainly chemical precipitation,and secondly surface physical adsorption.
active magnesium oxide;preparation;thorium;removal
2016-11-11
国家自然科学基金资助项目(11205030,11465002,41201500,11205031,41562011);江西省教育厅项目(GJJ14478);江西省教育厅科技落地计划项目(KJLD13054);核资源与环境省部共建国家重点实验室项目(NRE1320,Z201406)。
秦启凤(1993-),女,云南丽江人,硕士研究生,主要研究方向辐射防护与环境保护。
李小燕(1974-),女,甘肃镇原人,博士,副教授,主要研究方向为辐射防护与环境保护。E-mail:zhxz2004@163.com。
X591
A
1009-2617(2017)03-0208-05
10.13355/j.cnki.sfyj.2017.03.010
