Crosstalk of liver immune cells and cell death mechanisms in different murine models of liver injury and its clinical relevance
2017-06-19
Faisalabad, Pakistan
Crosstalk of liver immune cells and cell death mechanisms in different murine models of liver injury and its clinical relevance
Hilal Ahmad Khan, Muhammad Zishan Ahmad, Junaid Ali Khan and Muhammad Imran Arshad
Faisalabad, Pakistan
BACKGROUND: Liver inflammation or hepatitis is a result of pluripotent interactions of cell death molecules, cytokines, chemokines and the resident immune cells collectively called as microenvironment. The interplay of these inflammatory mediators and switching of immune responses during hepatotoxic, viral, drug-induced and immune cell-mediated hepatitis decide the fate of liver pathology. The present review aimed to describe the mechanisms of liver injury, its relevance to human liver pathology and insights for the future therapeutic interventions.
DATA SOURCES: The data of mouse hepatic models and relevant human liver diseases presented in this review are systematically collected from PubMed, ScienceDirect and the Web of Science databases published in English.
RESULTS: The hepatotoxic liver injury in mice induced by the metabolites of CCl4, acetaminophen or alcohol represent necrotic cell death with activation of cytochrome pathway, formation of reactive oxygen species (ROS) and mitochondrial damage. The Fas or TNF-α induced apoptotic liver injury was dependent on activation of caspases, release of cytochrome c and apoptosome formation. The ConA-hepatitis demonstrated the involvement of TRAIL-dependent necrotic/necroptotic cell death with activation of RIPK1/3. The α-GalCer-induced liver injury was mediated by TNF-α. The LPS-induced hepatitis involved TNF-α, Fas/FasL, and perforin/granzyme cell death pathways. The MHV3 or Poly(I:C) induced liver injury was mediated by natural killer cells and TNF-α signaling. The necrotic ischemia-reperfusion liver injury was mediated byhypoxia, ROS, and pro-in flammatory cytokines; however, necroptotic cell death was found in partial hepatectomy. The crucial role of immune cells and cell death mediators in viral hepatitis (HBV, HCV), drug-induced liver injury, non-alcoholic fatty liver disease and alcoholic liver disease in human were discussed.
CONCLUSIONS: The mouse animal models of hepatitis provide a parallel approach for the study of human liver pathology. Blocking or stimulating the pathways associated with liver cell death could unveil the novel therapeutic strategies in the management of liver diseases.
(Hepatobiliary Pancreat Dis Int 2017;16:245-256)
liver immunobiology; hepatitis; therapy; mode of cell death
Introduction
The liver is a pivotal organ of the body and plays a crucial role in taking up of nutrients from the gastrointestinal tract, storage of nutrients, metabolism, homeostasis, detoxification, immune regulation and tolerance, synthesis of bile, serum proteins, coagulating factors and complement proteins of the immune system. The liver is regarded as special immunological organ due to its enriched resident immune cell population like natural killer (NK) cells, NKT cells (formally called pit cells), Kupffer cells (KCs, resident macrophages of liver), dendritic cells (DCs), hepatic stellate cells (HSCs), liver sinusoidal endothelial cells (LSEC), innate lymphoid cells (ILCs), B cells, T cells and cells of myeloid lineage.[1]The ILCs are distinctly classified as ILC1s, ILC2s and ILC3s depending upon cytokine production and transcription factors involved in their development and function. ILC1s produce interferon-γ (IFN-γ) and they are dependent of transcription factor T-bet, ILC2s secrete Th2 cytokines such as IL-4, IL-5, IL-9, IL-13 andamphiregulin and require GATA3, and ILC3s produce IL-17 and/or IL-22 dependent on RORγt regulation.[2]The liver encounters many circulating antigens and toxins of gut origin. In such a precarious milieu, the liver must have a robust immunologic mechanism to deal with constant exposure to potential insult.
The liver is an organ with dual face immunological functions. On one hand, immune reactions against harmless antigens have to be avoided (immune tolerance), but on the other hand, to combat against the hepato-tropic infectious agents (viruses, bacteria, parasites), effective immune responses have to be induced.[3,4]The“dual edge” functions are regulated and controlled by the resident immune cells of the liver by secreting chemical mediators such as chemokines (for chemotaxis, recruitment of immune cells) and cytokines (pro-inflammatory and anti-inflammatory functions) collectively called as“microenvironment”. The pathophysiology of acute liver injury is orchestrated by the interplay of immune cells, cytokines, and parenchymal liver cells. The exacerbated immune responses following entry of antigens result in liver inflammation. Acute hepatitis is defined by liver cell death, cellular disarray and immune cells infiltration in the liver. The type of antigen, immunological reaction, cell death pathways or mode of liver cell death determines the fate of liver or immuno-pathogenesis of liver diseases (acute vs chronic) with distinct mechanisms of disease.
Animal models of hepatitis provide an excellent tool to understand the pathophysiological mechanisms and to correlate the data with clinical findings. The use of animal models in scientific research is appreciable due to mimicry with many human pathologies, easy availability of technical tools for analysis, reproducibility of data, close relevance to human parameters (physiology/metabolism), a minimal hazard to personnel and genetic deletion or insertion to study the effect of a specific gene. The animal models serve as an alternative approach for certain infectious diseases in human caused by HBV, HCV, influenza virus and HIV. Among various existent animal models of acute hepatitis, the present review summarized the mouse models of acute hepatitis with principal cellular and effective molecular players involved in liver cell death. The murine models of acute hepatitis have been categorized depending upon the nature of hepatitis inducing agent, i.e. hepatotoxin, autoimmune, immune cell dependent, Toll-like receptor (TLR) agonists, viral and fulminant hepatic models.
The hepatocytes express death receptors like Fas (CD95), TRAIL-R1 (DR4), TRAIL-R2 (DR5), TNFR1 and TNFR2 on their surface and the immune cells express death ligands like FasL, TRAIL, and TNF-α.[5]The interaction of these death ligands and receptors in different liver diseases lead to liver cell death (apoptosis, necrosis or necroptosis) and it determines the outcome of a disease. Briefly, the apoptosis is a highly organized and genetically controlled type of cell death mediated by distinct extrinsic (death receptor dependent) and intrinsic (mitochondrial/caspase dependent) pathways. The key features of the apoptotic mode of cell death are membrane blebbing, shrinkage of the cell, chromatin condensation, nuclear fragmentation and formation of apoptotic bodies.[6]Necrosis is characterized by oncosis (swelling) and the formation of plasma membrane blebs (devoid of organelles) and rupture of the plasma membrane[6]accompanied by a complete release of cellular constituents into the extracellular environment.
Evolving data[5-7]have evidenced involvement of a novel cell death pathway in liver pathology termed as necroptosis. The term “programmed necrosis” or“necroptosis” was described as an alternative receptor interacting protein kinase (RIPK) mediated form of cell death initiated by necrosis factor receptors, Fas and TNF-related apoptosis inducing ligand (TRAIL).[7]The necroptosis pathway is initiated by TNF receptors mainly dependent on RIPK1/RIPK3 activation and it is identified as “back up” cell death mechanism of apoptosis. Necroptosis is marked by cell and organelle swelling, extensive formation of intracellular vacuoles and rapid rupture of the plasma membrane.[6,8]The execution of necroptosis starts from binding and trimerization of death ligands (TNF-α, FasL, and TRAIL) to their cognate receptors. Briefly, the downstream events of TNF-induced necroptosis are initiated by the activation/trimerization of TNF and its receptor (TNFR1) that promotes the formation of complex I (containing signaling molecules TRADD, TRAF2, TRAF5, cIAP1, cIAP2 and RIP1) and complex II (caspase-8-dependent cleavage of RIPK1 and RIPK3). Moreover, the interplay of RIPK1, capase-8 and substrate of RIPK3 called as phosphorylated mixed lineage kinase domain-like (MLKL) defines the mode of cell death. The RIPK3-MLKL pathway (ubiquitylation) is essential to drive the necroptotic cell death while RIPK1-caspase-8 activation is required for the apoptotic cell death.[9]However, in the absence of caspase-8, RIPK1 stimulates necroptotic cell death.[9]It has been shown that necroptosis plays a crucial role in immune cell mediated hepatitis in mice because the inhibitors of necroptosis (i.e., necrostatin-1 and PJ34) protected liver injury in mice.[10,11]Necrostatin-1 (Nec-1), a small tryptophanbased compound, an inhibitor of RIPK1 activity (phosphorylation), blocks the interaction between RIPK1 and RIPK3 and inhibits necroptosis. Nec-1 is widely used in cellular and animal disease models to prevent the necroptotic cell death.[12]The present review comprehen-sively describes the cellular, molecular, immunological and cell death mechanisms of liver injury or hepatitis, its relevance with the human pathology and insights for novel therapeutic interventions.
Hepatotoxic murine liver injury models
Carbon tetrachloride (CCl4) induced acute hepatitisCCl4is a highly toxic liquid principally used in the manufacture of dichlorodi fluoromethane, it was used as refrigerant and propellant, and it is contained in fire extinguishers and in spot removers. However, CCl4exposure induced acute and chronic hepatitis. In mice, a single oral administration of CCl4triggers acute liver injury which is a widely studied model of acute hepatotoxicity. The hepatotoxic molecule CCl4activates cytochromes (CYP2E1, CYP2B1 or CYP2B2) to form trichloromethyl (CCl3) radical that reacts with oxygen to form a highly reactive oxygen species (ROS) that initiate lipid peroxidation and mitochondrial dependent liver injury and fatty degeneration.[13]The highly reactive species named as CCl3OO* starts lipid peroxidation and denaturation of polyunsaturated fatty acids. As a result, the mitochondrial, endoplasmic reticulum and plasma membrane permeability is lost with deregulation of Ca++in the cells leading to cellular demise.[13]Additionally, CCl4toxicity leads to hypomethylation of cellular components and liver damage.[13]
The increased in flux of cytokines, chemokines and immune cells like neutrophils following CCl4-induced liver injury result in hepatocyte damage (necrosis).[14]The IL-6 de ficient mice exhibited increased liver injury, in flammation, and delayed recovery following CCl4-induced liver injury compared to wild-type mice, suggesting a protective role of IL-6 via down-regulation of matrix metalloproteinase-2 (MMP-2).[15]The de ficiency of IL-10 led to more extensive CCl4-induced liver fibrosis and more prominent neutrophilic in filtration in IL-10 knockout mice during the acute CCl4challenge.[16]The neutrophils are crucial in this hepatic model and the liver invariant NKT (iNKT) cells have known to protect CCl4-induced hepatitis by limiting neutrophil in filtration.[14,17]The iNKT-de ficient mice (Jα-18 knockout) are more susceptible to CCl4-induced acute liver injury and in flammation[17,18]and the activation of iNKT cells by α-GalCer accelerates CCl4-induced acute liver injury, inlf ammation, and fibrosis.[18]The KCs play a vital role in CCl4-mediated hepatitis in mice as depletion of KCs protects CCl4-induced liver necrosis and IL-6 production.[19]Another study[20]demonstrated that CCl4-mediated hepatitis was dependent upon the activity of KCs via TNF-α and FasL. The de ficiency of chemokine receptor CCR6 in mice exacerbated the CCl4-induced liver in flammation with enhanced KCs recruitment.[21]Recently, it has been shown that chemical inhibition of c-Jun N-terminal kinase (JNK) 1 and 2 provided hepato-protection against CCl4-mediated hepatitis in mice.[22]
Acetaminophen/paracetamol induced acute hepatitis
Acetaminophen-induced hepatitis shares many features of hepatotoxic liver injury and administration of acetaminophen induces fulminant hepatitis with acute liver failure in mice.[23]In this model, mechanism of liver injury (necrosis) is dependent on the accumulation of acetaminophen metabolites formed by cytochrome P450, N-acetyl-p-benzoquinone imine (NAPQI), NAPQI protein adducts, glutathione depletion, oxidative stress, and mitochondrial damage.[24,25]The role of JNK1 and JNK2 was elaborated in acetaminophen-mediated liver injury as the use of chemical inhibitor (SP600125) of JNK protected mice against acetaminophen hepatitis.[22]
The in flammatory cytokines, such as TNF-α, IFN-γ, and IL-1β are crucial for the development of acetaminophen hepatitis.[26]The NK and NKT cells play a detrimental role in acetaminophen hepatitis as depletion of NK and NKT cell by anti-NK1.1 antibody protected mice against acetaminophen-induced liver injury.[27,28]The underlying liver injury was mediated by production of IFN-γ, chemokines, and up-regulation of FasL expression in the liver. A study demonstrated that the use of dimethyl sulfoxide (DMSO) to solubilize acetaminophen resulted in detrimental effect of NK and NKT cells in mice following acetaminophen hepatitis.[29]Indeed, the DMSO activated hepatic NK and NKT cellsin vivo, with increased NKT cell numbers and higher intracellular level of cytotoxic effector molecules like IFN-γ and granzyme B.[29]The necroptosis or programmed necrosis is involved in acetaminophen-mediated liver injury because blockade of either RIPK1 or RIPK3 was protective in acetaminophen liver injury.[30]
Alcohol induced acute hepatitis
The murine models of acute alcoholic hepatitis are widely used to correlate the findings with human pathology.[31]The experimental model of ethanol-induced liver injury represents a model of acute hepatitis and predominantly depends upon apoptotic liver damage.[32]Increased gut permeability to endotoxin leads to hypoxia-dependent liver injury[33]with induction of CYP2E1, cytochrome P450 isoforms, formation of ROS and lipid peroxides.[34]Furthermore, the release of pro-apoptotic factors such as cytochrome c into the cytosol and caspase activation leads to apoptotic liver injury in this model.[34]The immune molecules play an important role in alco-holic hepatitis as the alcohol-mediated hepatitis is associated with up-regulation of hepatic TLR9 and TNF-α activation.[35]The IL-22 was shown to induce hepatoprotective effects in alcohol-induced hepatotoxicity in mice with decrease in inflammation and promotion of anti-apoptotic activities.[36]
Cell death ligand mediated murine hepatic models
Anti-Jo2/FasL induced acute hepatitis
The liver is very sensitive to Fas mediated apoptosis because Fas receptor is constitutively expressed on hepatocytes. When mice are injected with anti-Fas antibody, death due to fulminant hepatitis and acute liver failure follows.[37,38]The administration of anti-Fas antibody rapidly induces severe damage to the liver (hepatocytes and sinusoidal endothelial cells) via massive apoptosis, indicating this animal model can be used in investigating human fulminant hepatitis.[37]The mode of apoptotic liver injury in this model largely depends upon activation of caspase-3 and caspase-7, release of cytochrome c and apoptosome formation.[39]Following injection of Jo2, it directly binds to Fas receptor in liver with CXC chemokine formation and in flammation dependent on caspases/mitochondrial damage and the transcription factor activator protein 1 (AP-1).[40,41]
TNF-α/D-galactosamine (D-GalN) induced acute hepatitis
Acute hepatitis induced by administration of TNF-α/ D-GalN represents an apoptotic model of hepatitis and death of mice occurs due to enhanced systemic shock and liver insuf ficiency. Indeed, the amino sugar GalN sensitizes the host when it is metabolized in the liver and results in selective depletion of uridine nucleotides, which speci fically inhibits transcription at hepatocyte level.[42]De ficiency of TNFR1 in mice makes them resistant to TNF-α/D-GalN treatment, demonstrating an essential role for TNFR1 in this apoptosis model.[42]However, the TNFR2 de ficient mice are more susceptible to TNF-α/D-GalN-induced liver injury suggesting that in the absence of TNFR2, more TNF-α is available to bind TNFR1 to enhance apoptosis.[42]The toxicity in the murine TNF-α model resembles viral form of acute hepatic failure in patients characterized by massive hepatocyte apoptosis via engagement of TNF receptors and downstream caspase-dependent liver injury.[25,43]
Immune cell mediated murine hepatic models
Concanavalin A (ConA)-induced acute hepatitis
ConA-induced hepatitis is a T-cell driven liver injury model and its features resemble with viral or autoimmune hepatitis in human.[35,44,45]ConA is a lectin, isolated from jack bean (Canavalia brasiliensis), it binds to mannose residues (α-D-mannoside, methyl-α-D-mannopyranoside, α-D-glucose, and methyl-α-D-glucose) of different glycoproteins and thereby activates lymphocytes. The ConA-induced murine hepatic model was first developed by Tiegs and colleagues in 1992.[44]Upon a single intravenous injection to mice, ConA induces acute liver damage within 8 hours in a dose range from 10-25 mg/kg. It has been shown that 15 minutes after intravenous administration of ConA, it binds to LSEC, KCs, CD4+ T cells and NKT cells to induce inflammation and hepatocyte damage.[46]ConA induces hepatocyte death by stimulation of CD4+ T cells, NKT cells and KCs in liver, resulting in secretion of copious amounts of proinflammatory cytokines like TNF-α, IFN-γ, IL-6, IL-12, IL-18, and chemokines.[35,44,45]In addition, the IL-10 is considered to be an anti-inflammatory cytokine and have a protective effect in this model.[47]
The necrotic death of hepatocytes induced by ConA is accompanied by release of the aminotransferases (ALT and AST) from the cytoplasm of hepatocytes into the blood, inflammatory infiltration into the liver consisting of neutrophils, macrophages and T cells.[35,44,45]The cytotoxic effector molecules and their receptors play a key role in the development of ConA-induced liver cell death. Among these molecules, the perforin-granzyme system is essential for ConA hepatitis along with induction of intracellular adhesion molecules as well as influx of IFN-γ.[48]In ConA-induced hepatitis, the production of large amounts of IFN-γ by activated T cells is essential for liver injury,[35,45]and ConA hepatitis is suppressed in IFN-γ deficient mice, explaining its critical contribution in this model.[49]A crucial role for TNF-α and FasL/Fas in ConA hepatitis is evident from previous studies[50]because anti-TNF-α antibodies inhibited hepatitis in this model.[51]
Evolving data has implied the role of TRAIL and its receptor (death receptor 5, DR5) in liver diseases in mice by the use of recombinant TRAIL or agonist anti-DR5-antibody in murine hepatitis.[52-54]The TRAIL is expressed by myeloid or lymphoid immune cells in the liver like NK and NKT cells while DR5 expression is mainly found on hepatocytes. Expression of TRAIL and DR5 is increased following ConA hepatitis evidencing a critical contribution of these molecules to the development of hepatitis. The study[52]demonstrated the critical role of TRAIL in ConA hepatitis because liver cell death was suppressed in TRAIL-knockout mice or by blocking of DR5 receptor. In human, TRAIL interacts with four receptors i.e. TRAIL-R1 (death receptor 4), TRAIL-R2(death receptor 5, KILLER, or TRICK-2), TRAIL-R3 (DcR1) and TRAIL-R4 (DcR2), however, TRAIL-R1 and TRAIL-R2 induce apoptosis while TRAIL-R3 and TRAIL-R4 could not induce apoptosis.[55,56]In mice, only one TRAIL receptor (DR5) has been identi fied that shares homology with human DR5 or TRAIL-R2 and activates cell death.[57]The STAT4 was proved to be hepatoprotective during ConA hepatitis in mice as genetic ablation of STAT4 in mice exhibited enhanced liver injury.[58]
During ConA hepatitis, the downstream cascade of cell death signaling and adaptor protein complexes after engagement of cell death ligands and receptors result in necrotic and necroptotic liver cell death. Recent findings demonstrated the involvement of necroptotic cell death pathway in ConA hepatitis and pre-treatment of mice with Nec-1 and PJ34 (inhibitors of RIPK1 and PARP-1) ameliorated ConA-induced liver injury.[10,11]These studies suggested that inhibition of necroptotic cell death pathway may have therapeutic potential in the treatment of immune cell mediated hepatitis.
α-Galactosylceramide (α-GalCer)-induced acute hepatitis
The α-GalCer is a glycolipid, originally isolated from a marine sponge (Sphingomonas microorganism), which speci fically activates iNKT cells (Vα14 NKT cells) via antigen presenting cells in the context of CD1d. The α-GalCer-induced hepatitis is associated with up-regulated expression of pro-in flammatory cytokines like TNF-α, IFN-γ, IL-2, IL-4, and IL-6 produced by activated NKT cells in the liver.[59,60]However, the acute liver injury induced by α-GalCer administration in mice is mediated by TNF-α and independent of KCs.[60]Pre-treatment of mice with D-GalN exacerbated the α-GalCer mediated hepatitis in mice with massive parenchymal hemorrhage, hepatocyte apoptosis and sinusoidal endothelial cell injury.[61]Recently, it is reported that the liver iNKT cells produce IL-17 in response to α-GalCer stimulation which induce protective effect on α-GalCer liver injury.[62]Depletion of IL-17 by neutralizing antibodies aggravates the α-GalCer-induced liver injury, with increased hepatic neutrophil and monocyte in filtration.[62]The administration of recombinant IL-17 abolishes these effects.[62]The role of resident hepatic B cells was dissected in α-GalCer mediated liver injury demonstrating that iNKT cells stimulation and recruitment of innate-like regulatory B cells to the liver suppressed the liver in flammation.[63]
TLR3 agonist, Poly(I:C) induced acute hepatitis
The polyinosine-polycytidylic acid Poly(I:C) is a synthetic analog of double-stranded RNA (dsRNA), a molecular pattern associated with viral infections. The Poly(I:C) is a viral dsRNA mimetic, sensed by the endosomal receptor, TLR3,[64]as well as recently discovered cytoplasmic receptors, such as RNA helicase retinoic acid-inducible gene-I (RIGI) and melanoma differentiation-associate gene 5 (MDA-5).[65]Upon Poly(I:C) recognition by the immune cells, TLR3 activates the transcription factor interferon regulatory factor 3 (IRF3), through the adapter protein Toll-IL-1 receptor (TIR) domain containing adapter inducing IFN-α (TRIF, also known as TICAM-1).[66]Activation of IRF3 leads to the production of type I IFN, especially IFN-β. A second pathway involves the recruitment of TNF receptor-associated factor 6 (TRAF6) or receptor interacting protein 1 (RIP1), with the subsequent activation of transcription factors NF-κB and AP-1.[67]
Poly(I:C) activates macrophages, NK cells, and other lymphocyte sub-population[68]with induction of in flammatory cytokines (TNF-α, IFN-γ, IL-6, and IL-12) and Type I IFN (IFN-α and IFN-β), a similar signature found during viral hepatitis.[64,69,70]The Poly(I:C) administration in mice induces acute liver injury and the liver injury becomes lethal or aggravates if mice are pre-treated with D-GalN.[70]The NK cells mediated acute hepatitis by Poly(I:C) has been shown to be attenuated in Bruton’ s tyrosine kinase (Btk) knockout mice, implicating a role for this kinase in TLR3 dependent liver injury and NK cells activation in the liver.[71]The Poly(I:C) treatment induced production of IL-17A from hepatic γδT cells which aggravated the liver injury in mice suggesting a detrimental or pathological role of IL-17 in Poly(I: C)-induced hepatitis.[72]Our data demonstrated upregulated expression of IL-33 during Poly(I:C) fulminant hepatitis[73]and the hepatocyte-speci fic IL-33 expression was down-regulated by treatment of PJ34.[11]Moreover, a protective role of regulatory T cells (Treg) was found in Poly(I:C)-induced hepatitis that was dependent upon production of inhibitory cytokines (TGF-β and IL-10) in the liver.[74]
Bacterial toxin (or LPS) induced acute hepatitis
The lipopolysaccharides (LPS) are major pathogenic factors of Gram-negative bacteria that induce systemic pro-in flammatory responses culminating in multiple organ failure and death. The LPS/D-GalN-induced hepatitis in mice is a well known animal model of acute hepatic failure. In this model, the liver injury critically depends on macrophage (KCs) derived pro-in flammatory cytokines, including IL-1, IL-6, and TNF-α.[75]The soluble TNF-α (but not membrane TNF-α) mediates LPS-induced hepatitis in mice.[76]The D-GalN/LPS induced liver injury was dependent on neutrophil activation and TNF-α production which caused hepatocyte necro-sis and organ failure.[77]Other studies had shown that TNF-α mediated caspase-dependent hepatocyte apoptosis as a predominant mechanism of LPS associated liver injury.[75,78]The hepatocyte apoptosis in D-GalN/LPS induced hepatitis is mediated not only by TNF-α and Fas/FasL cytotoxicity but also involves a perforin/granzyme cell death pathway.[79]Interestingly, the activation of TLR3 ligand by Poly(I:C) attenuated the LPS/D-GalN-induced fulminant hepatitis by down-regulation of TLR4 expression in liver macrophages (KCs).[75]The bacterial toxin such as Pseudomonas aeruginosa exotoxin A (PEA) induces liver injury via activation of NK cells, T cells and KCs in association with increased expression of IFN-γ, TNF-α, IL-18 and perforin.[80,81]Recent studies showed that the LPS/D-GalN-induced liver injury employed the hepatocyte intrinsic TNFR1 pathway in mice following secretion of TNF-α by activated KCs in liver.[82,83]The cytokine IL-17A played a regulatory role in neutrophil induced liver injury following LPS/D-GalN injection as the in flammatory response was decreased and the survival rate was increased in IL-17A de ficient mice compared to wild-type mice.[84]The pharmacological inhibition of phosphoinositide 3-kinase (PI3K) led to hepato-protection in LPS-injected mice by suppressing the phosphorylation of IκB.[85]
Mouse hepatitis virus type 3 (MHV3) induced acute hepatitis
The coronaviruses, including MHV are large, enveloped, positive-strand RNA viruses, with genome ranging in size from 27-32 kb. The hepatotropic MHV3 serotype induced severe fulminant hepatitis in mice with lethality depending upon virus strain, route of infection, age, genetic background and immune status of the mice.[86]Several strains of MHV induce acute encephalitis and acute and chronic demyelinating disease in mice.[86,87]The MHV-induced hepatitis is an excellent model for studying the immunological disorders associated with viral hepatitis and it has mimicry with human HBV infection.
The MHV interacts with a speci fic receptor called carcino embryonic cell adhesion antigen 1 (CEACAM1) which is expressed by the hepatocytes, LSEC, NK cells, and KCs.[88]The MHV3 (pathogenic strain L2-MHV3) can replicate in the hepatocytes, LSEC and KCs, leading to virus induced necrotic cell death.[88]The pathogenic L2-MHV3 virus is a cloned sub-strain isolated from the liver of infected DBA2 mice and propagated in L2 cells (continuous mouse fibroblast L2 cell line). The pathogenic L2-MHV3 virus induces fulminant hepatitis via activation of NK cells in susceptible C57BL/6 mice and their death within 3-5 days post-infection with extensive necrosis in the liver and immunode ficiency in several lymphoid organs.[88-90]Another pathogenic strain, MHV-A59 (derived from normal mouse liver cell line-NCTC-1469 or from L2 cells) induced fulminant hepatitis as well as autoimmunity in susceptible C57BL/6 mice with formation of autoantibodies (autoAb) to fumarylacetoacetate hydrolase (FAH), a soluble cytosolic enzyme present in the liver and kidneys.[91]The acute liver injury (after 3-5 days infection) induced by MHV-A59 is associated with an increase level of IL-6, TNF-α and IL-17 in mice.[89]In contrast, the non-pathogenic strain isolated from persistently infected YAC lymphoid cell line (YACMHV3) does not induce an acute lethal disease but only subclinical infection in mice.[92]Recent data evidenced the invasion of MHV in the brain microvasculature by impairment of IFN-β production[93]or by sustained CXCL1 expression.[94]The pathogenic infection by MHV3 in mice up-regulated expression of IL-33 in the liver along with other pro-inflammatory cytokines such as IL-6, TNF-α, IL-1β, and IFN-γ.[73]
In summary, the hepatotoxic agents such as CCl4, acetaminophen, alcohol or their metabolites, the viral infectious agent (MHV3) or TLR-mimetic LPS or Poly(I: C), the death ligands (Fas, TRAIL or TNF-α) and the immune-cells activating hepato-tropic agents like ConA/ α-GalCer interact with specific resident immune cells in the liver. The interplay of invading hepato-tropic antigens with NK cells, NKT cells, ILCs, KCs, LSECs, vascular endothelial cells (VECs), and DCs results in release of cytokine or inflammatory mediators that lead to liver injury via apoptosis, necrosis or necroptosis (Fig. 1) depending upon the agent involved (Table). The liver cell death following receptor-mediated recognition of hepatotropic agents is initiated by the engagement of cell death ligands such as Fas, TRAIL, and TNF-α with their cognate receptors FasR, TRAIL-R, and TNF-R, respectively. The cell death signaling is represented by the formation of cell death platforms called as complex-I and complex-II importantly comprising of adaptor molecules such as FADD, RIPK1, RIPK3 caspase-8, and TRAD. The downstream signaling is the execution of mode of cell death by the action of caspases (apoptosis), RIPK1-FADD-TRAD pathway (necrosis) or RIPK1-RIPK3-FADD pathway (necroptosis) with subsequent activation of transcription factors (Fig. 2).
Surgical models of mouse hepatitis and mode of liver cell death
The necrotic mode of cell death is implicated during surgically induced liver injury such as ischemia-reperfusion (I/R), bile duct ligation (BDL) and partial hepatectomy.[5,95]During these liver injury models, hypoxia leadsto depletion of ATP and loss of cell viability by necrotic cell death.[5]The liver injury is also associated with simultaneous activation of KCs, release of ROS, pro-inflammatory cytokines, and chemokines that participate in liver cell death.[95]Moreover, the hepatocytes undergo necroptotic cell death during partial hepatectomy[96]with induction of RIPK3 expression in hepatocytes. In partial hepatectomy, the regeneration of the liver is the main feature controlled by immune mediators with apoptosis and autophagy as dominant modes of cell death in this model.[97]The surgical mouse hepatic models may represent the mechanism of liver injury followed by livertransplantation or liver surgery in human.

Fig. 1. Role of immune cells and cytokine mediators in the progression of acute hepatitis. The invading hepato-tropic antigens (toxic and infectious) are recognized by the immune cells of the liver through pattern recognition receptors which initiate the liver injury or hepatitis. The crosstalk of antigens with host cells, exacerbated immune response and mode of cell death lead to the development of liver injury.
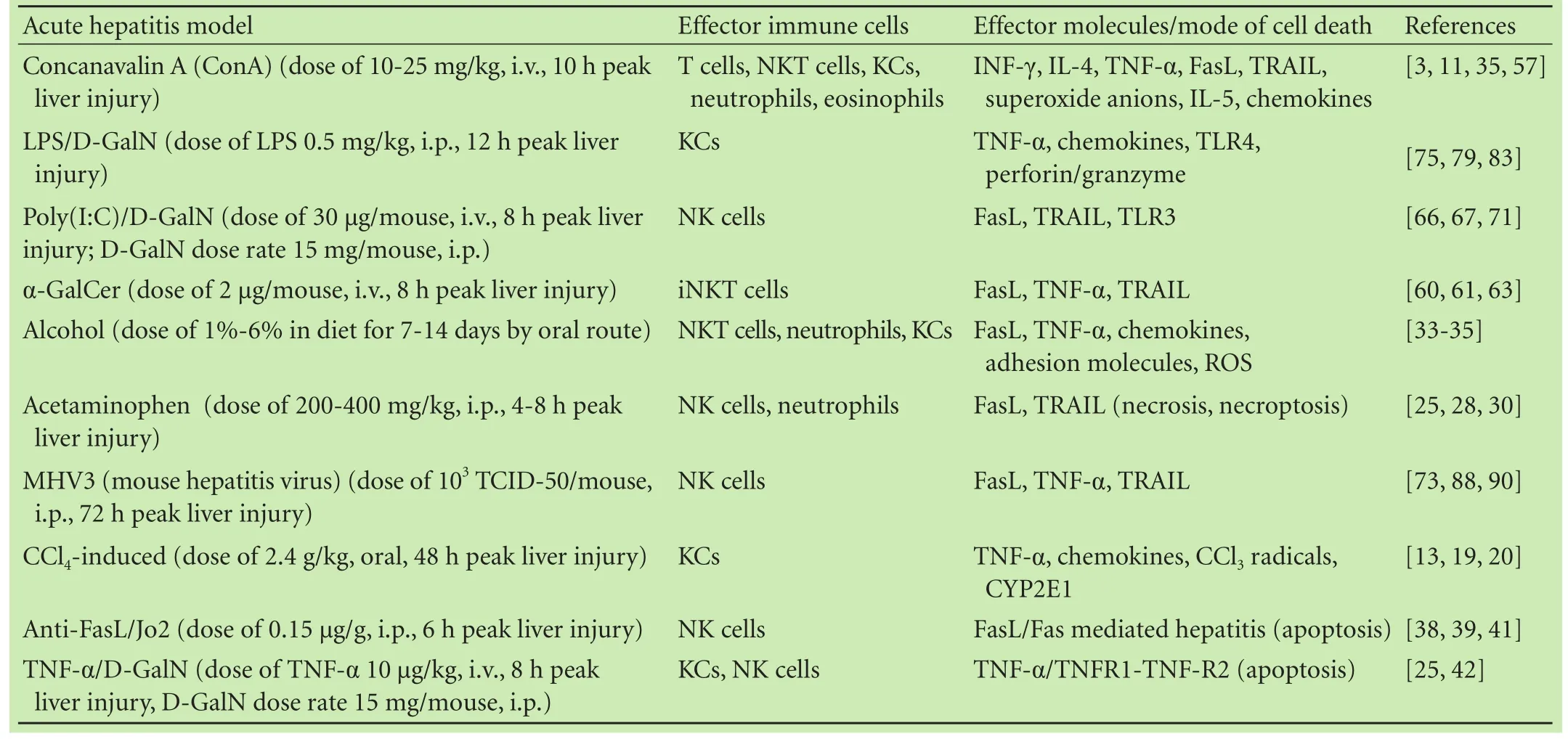
Table. In vivo murine models of acute hepatitis and involvement of immune or cell death mediators

Fig. 2. Mode of liver cell death and signaling pathway. Mechanism of liver injury induced by CCl4, acetaminophen, alcohol, ConA, α-Galcer, Poly(I:C) and LPS (endotoxins), TNF-α, TNF-related apoptosis-inducing ligand (TRAIL) and Fas ligand (FasL). Pro-inflammatory cytokines are produced by many liver cell types including KCs, NK cells, NKT cells and lymphocytes in response to inflammation, infection and other environmental stresses, which in turn cause liver injury via different modes of cell death importantly apoptosis, necrosis and necroptosis.
Relevance of liver cell death mechanisms with human pathology
Cell death in the liver is essentially a result of chronic derangement of liver homeostasis[98]which accounts for a large number of chronic diseases of the liver. Programmed cell death is a key homeostatic component as it gets rid of chronically ill cells before they become malignant.[98]The KCs secrete TNF-α, NKT cells express FasL and NK cells express TRAIL; these immune cells are principal mediators of cell death in human liver pathology.[99]Briefly, in viral hepatitis, there is a direct relationship between apoptosis and inflammation such as in HCV infection[100]with enhanced activation of caspases in ongoing viral inflammation.[101]In HCV infection, NK cells are activated by type I IFN and DCs[102]to produce TNF-α and liver damage. Apoptosis stimulated by FasL provides an efficient mean to remove unwanted HBV/HCV-infected hepatocytes and liver cancer cells by T lymphocytes.[5]Viral hepatitis by HBV and HCV increases Fas expression on hepatocytes and TRAIL expression on NK cells for the elimination of viruses.[103]In response to chronic liver disease, NK cells express apoptosis-inducing mediators such as TRAIL-R2 and Fas that drive apoptosis in HBV infection in human.[104]The TNF-α and TNFR1 have also been implicated in driving apoptosis in HCV via cytotoxic T lymphocytes.[5,101]
In drug-induced liver injury which is idiosyncratic drug-induced liver injury with certain haplotype human leukocyte antigen (HLA) genetic predisposition has been linked to immune-mediated apoptosis. Hapten presentation leads to activation of cytotoxic CD8 T-cells with the expression of FasL, TNF-α and to a smaller extent perforins that mediate cell death.[105]
In non-alcoholic fatty liver disease (NAFLD), the immune cells like monocytes and macrophages play an important role in liver injury. These cells secrete inflammatory cytokines IL-6 and TNF-α to aggravate the liver damage.[106]In severe form of NAFLD, namely nonalcoholic steatohepatitis (NASH), in a milieu of exacerbated inflammation and fibrosis, expression of death receptors such as Fas, TRAILR-2, and TNF receptor is increased.[107]The Fas expression and infiltration of FasL-expressing cytotoxic T lymphocytes led to an apoptotic liver injury. Recent data correlated the human and murine models of NASH and demonstrated the over-expression of RIPK3 in human NASH and in a dietary mouse model of steatohepatitis.[108]The underlying mechanism was shown to be mediated by RIPK3 and JNK necrop-tosis signaling, the release of inflammatory mediators, liver infiltration of macrophages culminating in liver cell death and fibrosis.[108]Studies in the past have reported the role of polymorphonuclear cells in cell death during alcoholic liver disease in human[109,110]with increased expression of Fas and TNFR1.[106]However, more recent literature implicates a greater role of KCs and TNF-α as the main inflammatory mediator that activates the apoptotic pathway.[107]The above data suggested that the cell death pathways and immune cells play a vital role in the human liver pathology and there is mimicry of mechanisms of liver disease with murine models of hepatitis.
Conclusions
The mechanisms of liver cell death and the crucial role of immune mediators in liver pathology in different animal models of hepatitis will provide the basis for the understanding of human liver disease or its relevance to clinical pathology. Modulation of immune cells-mediated liver injury and targeting of liver cell death pathways by chemical inhibitors could be promising strategies for the treatment of liver diseases. In the future, studies focusing on novel therapeutic targets or interventions in mouse hepatic models will be needed to translate the findings into clinical practice.
Contributors:AMI proposed the study. KHA and AMI wrote the first draft. KHA collected and analyzed the bibliography. KJA helped in writing of liver physiology and figures. AMZ contributed in experimental trials. AMI is the guarantor.
Funding:This study was supported by a grant from Higher Education Commission (HEC) at University of Agriculture, Faisalabad, Pakistan (No. 20-4613/NRPU/R&D/HEC/14/45).
Ethical approval:Not needed.
Competing interest:The authors do not choose to declare any conflict of interest related directly or indirectly to the subject of this article.
1 Heymann F, Tacke F. Immunology in the liver--from homeostasis to disease. Nat Rev Gastroenterol Hepatol 2016;13:88-110.
2 Sonnenberg GF, Artis D. Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nat Med 2015;21:698-708.
3 Erhardt A, Tiegs G. Tolerance induction in response to liver inflammation. Dig Dis 2010;28:86-92.
4 Karimi MH, Geramizadeh B, Malek-Hosseini SA. Tolerance Induction in Liver. Int J Organ Transplant Med 2015;6:45-54.
5 Malhi H, Guicciardi ME, Gores GJ. Hepatocyte death: a clear and present danger. Physiol Rev 2010;90:1165-1194.
6 Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 2012;19:107-120.
7 Chan FK, Shisler J, Bixby JG, Felices M, Zheng L, Appel M, et al. A role for tumor necrosis factor receptor-2 and receptorinteracting protein in programmed necrosis and antiviral responses. J Biol Chem 2003;278:51613-51621.
8 Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 2010;11:700-714.
9 Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D’Cruz AA, et al. RIPK3 promotes cell death and NLRP3 in flammasome activation in the absence of MLKL. Nat Commun 2015;6:6282.
10 Jouan-Lanhouet S, Arshad MI, Piquet-Pellorce C, Martin-Chouly C, Le Moigne-Muller G, Van Herreweghe F, et al. TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ 2012;19:2003-2014.
11 Arshad MI, Piquet-Pellorce C, Filliol A, L’Helgoualc’h A, Lucas-Clerc C, Jouan-Lanhouet S, et al. The chemical inhibitors of cellular death, PJ34 and Necrostatin-1, down-regulate IL-33 expression in liver. J Mol Med (Berl) 2015;93:867-878.
12 Smith CC, Yellon DM. Necroptosis, necrostatins and tissue injury. J Cell Mol Med 2011;15:1797-1806.
13 Weber LW, Boll M, Stamp flA. Hepatotoxicity and mechanism of action of haloalkanes: carbon tetrachloride as a toxicological model. Crit Rev Toxicol 2003;33:105-136.
14 Ramaiah SK, Jaeschke H. Role of neutrophils in the pathogenesis of acute in flammatory liver injury. Toxicol Pathol 2007;35:757-766.
15 Bansal MB, Kovalovich K, Gupta R, Li W, Agarwal A, Radbill B, et al. Interleukin-6 protects hepatocytes from CCl4-mediated necrosis and apoptosis in mice by reducing MMP-2 expression. J Hepatol 2005;42:548-556.
16 Louis H, Van Laethem JL, Wu W, Quertinmont E, Degraef C, Van den Berg K, et al. Interleukin-10 controls neutrophilic in filtration, hepatocyte proliferation, and liver fibrosis induced by carbon tetrachloride in mice. Hepatology 1998;28:1607-1615.
17 Lisbonne M, L’Helgoualc’h A, Nauwelaers G, Turlin B, Lucas C, Herbelin A, et al. Invariant natural killer T-cell-de ficient mice display increased CCl4-induced hepatitis associated with CXCL1 over-expression and neutrophil in filtration. Eur J Immunol 2011;41:1720-1732.
18 Park O, Jeong WI, Wang L, Wang H, Lian ZX, Gershwin ME, et al. Diverse roles of invariant natural killer T cells in liver injury and fibrosis induced by carbon tetrachloride. Hepatology 2009;49:1683-1694.
19 Kiso K, Ueno S, Fukuda M, Ichi I, Kobayashi K, Sakai T, et al. The role of Kupffer cells in carbon tetrachloride intoxication in mice. Biol Pharm Bull 2012;35:980-983.
20 Sato A, Nakashima H, Nakashima M, Ikarashi M, Nishiyama K, Kinoshita M, et al. Involvement of the TNF and FasL produced by CD11b Kupffer cells/macrophages in CCl4-induced acute hepatic injury. PLoS One 2014;9:e92515.
21 Affò S, Rodrigo-Torres D, Blaya D, Morales-Ibanez O, Coll M, Millán C, et al. Chemokine receptor Ccr6 de ficiency alters hepatic in flammatory cell recruitment and promotes liver inflammation and fibrosis. PLoS One 2015;10:e0145147.
22 Cubero FJ, Zoubek ME, Hu W, Peng J, Zhao G, Nevzorova YA, et al. Combined activities of JNK1 and JNK2 in hepatocytes protect against toxic liver injury. Gastroenterology2016;150:968-981.
23 Imaeda AB, Watanabe A, Sohail MA, Mahmood S, Mohamadnejad M, Sutterwala FS, et al. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J Clin Invest 2009;119:305-314.
24 Larson AM. Acetaminophen hepatotoxicity. Clin Liver Dis 2007;11:525-548, vi.
25 Bantel H, Schulze-Osthoff K. Mechanisms of cell death in acute liver failure. Front Physiol 2012;3:79.
26 Ishida Y, Kondo T, Ohshima T, Fujiwara H, Iwakura Y, Mukaida N. A pivotal involvement of IFN-gamma in the pathogenesis of acetaminophen-induced acute liver injury. FASEB J 2002;16:1227-1236.
27 Liu ZX, Govindarajan S, Kaplowitz N. Innate immune system plays a critical role in determining the progression and severity of acetaminophen hepatotoxicity. Gastroenterology 2004;127:1760-1774.
28 Cover C, Liu J, Farhood A, Malle E, Waalkes MP, Bajt ML, et al. Pathophysiological role of the acute inflammatory response during acetaminophen hepatotoxicity. Toxicol Appl Pharmacol 2006;216:98-107.
29 Masson MJ, Carpenter LD, Graf ML, Pohl LR. Pathogenic role of natural killer T and natural killer cells in acetaminopheninduced liver injury in mice is dependent on the presence of dimethyl sulfoxide. Hepatology 2008;48:889-897.
30 Deutsch M, Graffeo CS, Rokosh R, Pansari M, Ochi A, Levie EM, et al. Divergent effects of RIP1 or RIP3 blockade in murine models of acute liver injury. Cell Death Dis 2015;6:e1759.
31 Wilkin RJ, Lalor PF, Parker R, Newsome PN. Murine models of acute alcoholic hepatitis and their relevance to human disease. Am J Pathol 2016;186:748-760.
32 Benedetti A, Brunelli E, Risicato R, Cilluffo T, Jézéquel AM, Orlandi F. Subcellular changes and apoptosis induced by ethanol in rat liver. J Hepatol 1988;6:137-143.
33 Tilg H, Day CP. Management strategies in alcoholic liver disease. Nat Clin Pract Gastroenterol Hepatol 2007;4:24-34.
34 Guicciardi ME, Gores GJ. Apoptosis: a mechanism of acute and chronic liver injury. Gut 2005;54:1024-1033.
35 Wang HX, Liu M, Weng SY, Li JJ, Xie C, He HL, et al. Immune mechanisms of Concanavalin A model of autoimmune hepatitis. World J Gastroenterol 2012;18:119-125.
36 Xing WW, Zou MJ, Liu S, Xu T, Wang JX, Xu DG. Interleukin-22 protects against acute alcohol-induced hepatotoxicity in mice. Biosci Biotechnol Biochem 2011;75:1290-1294.
37 Ogasawara J, Watanabe-Fukunaga R, Adachi M, Matsuzawa A, Kasugai T, Kitamura Y, et al. Lethal effect of the anti-Fas antibody in mice. Nature 1993;364:806-809.
38 Haga S, Terui K, Zhang HQ, Enosawa S, Ogawa W, Inoue H, et al. Stat3 protects against Fas-induced liver injury by redoxdependent and -independent mechanisms. J Clin Invest 2003;112:989-998.
39 Bajt ML, Lawson JA, Vonderfecht SL, Gujral JS, Jaeschke H. Protection against Fas receptor-mediated apoptosis in hepatocytes and nonparenchymal cells by a caspase-8 inhibitor in vivo: evidence for a postmitochondrial processing of caspase-8. Toxicol Sci 2000;58:109-117.
40 Faouzi S, Burckhardt BE, Hanson JC, Campe CB, Schrum LW, Rippe RA, et al. Anti-Fas induces hepatic chemokines and promotes inflammation by an NF-kappa B-independent, caspase-3-dependent pathway. J Biol Chem 2001;276:49077-49082.
41 Walter D, Schmich K, Vogel S, Pick R, Kaufmann T, Hochmuth FC, et al. Switch from type II to I Fas/CD95 death signaling on in vitro culturing of primary hepatocytes. Hepatology 2008;48:1942-1953.
42 Bradham CA, Plümpe J, Manns MP, Brenner DA, Trautwein C. Mechanisms of hepatic toxicity. I. TNF-induced liver injury. Am J Physiol 1998;275:G387-392.
43 Sass G, Heinlein S, Agli A, Bang R, Schümann J, Tiegs G. Cytokine expression in three mouse models of experimental hepatitis. Cytokine 2002;19:115-120.
44 Tiegs G, Hentschel J, Wendel A. A T cell-dependent experimental liver injury in mice inducible by concanavalin A. J Clin Invest 1992;90:196-203.
45 Takeda K, Hayakawa Y, Van Kaer L, Matsuda H, Yagita H, Okumura K. Critical contribution of liver natural killer T cells to a murine model of hepatitis. Proc Natl Acad Sci U S A 2000;97:5498-5503.
46 Knolle PA, Gerken G, Loser E, Dienes HP, Gantner F, Tiegs G, et al. Role of sinusoidal endothelial cells of the liver in concanavalin A-induced hepatic injury in mice. Hepatology 1996;24:824-829.
47 Di Marco R, Xiang M, Zaccone P, Leonardi C, Franco S, Meroni P, et al. Concanavalin A-induced hepatitis in mice is prevented by interleukin (IL)-10 and exacerbated by endogenous IL-10 deficiency. Autoimmunity 1999;31:75-83.
48 Watanabe Y, Morita M, Akaike T. Concanavalin A induces perforin-mediated but not Fas-mediated hepatic injury. Hepatology 1996;24:702-710.
49 Tagawa Y, Sekikawa K, Iwakura Y. Suppression of concanavalin A-induced hepatitis in IFN-gamma(-/-) mice, but not in TNF-alpha(-/-) mice: role for IFN-gamma in activating apoptosis of hepatocytes. J Immunol 1997;159:1418-1428.
50 Schümann J, Wolf D, Pahl A, Brune K, Papadopoulos T, van Rooijen N, et al. Importance of Kupffer cells for T-cell-dependent liver injury in mice. Am J Pathol 2000;157:1671-1683.
51 Mizuhara H, O’Neill E, Seki N, Ogawa T, Kusunoki C, Otsuka K, et al. T cell activation-associated hepatic injury: mediation by tumor necrosis factors and protection by interleukin 6. J Exp Med 1994;179:1529-1537.
52 Zheng SJ, Wang P, Tsabary G, Chen YH. Critical roles of TRAIL in hepatic cell death and hepatic inflammation. J Clin Invest 2004;113:58-64.
53 Takeda K, Kojima Y, Ikejima K, Harada K, Yamashina S, Okumura K, et al. Death receptor 5 mediated-apoptosis contributes to cholestatic liver disease. Proc Natl Acad Sci U S A 2008;105:10895-10900.
54 Beraza N, Malato Y, Sander LE, Al-Masaoudi M, Freimuth J, Riethmacher D, et al. Hepatocyte-specific NEMO deletion promotes NK/NKT cell- and TRAIL-dependent liver damage. J Exp Med 2009;206:1727-1737.
55 Schneider P, Thome M, Burns K, Bodmer JL, Hofmann K, Kataoka T, et al. TRAIL receptors 1 (DR4) and 2 (DR5) signal FADD-dependent apoptosis and activate NF-kappaB. Immunity 1997;7:831-836.
56 Degli-Esposti MA, Dougall WC, Smolak PJ, Waugh JY, Smith CA, Goodwin RG. The novel receptor TRAIL-R4 induces NF-kappaB and protects against TRAIL-mediated apoptosis, yet retains an incomplete death domain. Immunity 1997;7:813-820.
57 Wu GS, Burns TF, Zhan Y, Alnemri ES, El-Deiry WS. Molecular cloning and functional analysis of the mouse homologue of the KILLER/DR5 tumor necrosis factor-relatedapoptosis-inducing ligand (TRAIL) death receptor. Cancer Res 1999;59:2770-2775.
58 Wang Y, Feng D, Wang H, Xu MJ, Park O, Li Y, et al. STAT4 knockout mice are more susceptible to concanavalin A-induced T-cell hepatitis. Am J Pathol 2014;184:1785-1794.
59 Leite-de-Moraes MC, Lisbonne M, Arnould A, Machavoine F, Herbelin A, Dy M, et al. Ligand-activated natural killer T lymphocytes promptly produce IL-3 and GM-CSF in vivo: relevance to peripheral myeloid recruitment. Eur J Immunol 2002;32:1897-1904.
60 Biburger M, Tiegs G. Alpha-galactosylceramide-induced liver injury in mice is mediated by TNF-alpha but independent of Kupffer cells. J Immunol 2005;175:1540-1550.
61 Fujii H, Seki S, Kobayashi S, Kitada T, Kawakita N, Adachi K, et al. A murine model of NKT cell-mediated liver injury induced by alpha-galactosylceramide/d-galactosamine. Virchows Arch 2005;446:663-673.
62 Wondimu Z, Santodomingo-Garzon T, Le T, Swain MG. Protective role of interleukin-17 in murine NKT cell-driven acute experimental hepatitis. Am J Pathol 2010;177:2334-2346.
63 Almishri W, Deans J, Swain MG. Rapid activation and hepatic recruitment of innate-like regulatory B cells after invariant NKT cell stimulation in mice. J Hepatol 2015;63:943-951.
64 Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Tolllike receptor 3. Nature 2001;413:732-738.
65 Takeuchi O, Akira S. Recognition of viruses by innate immunity. Immunol Rev 2007;220:214-224.
66 Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, et al. Role of adaptor TRIF in the MyD88-independent tolllike receptor signaling pathway. Science 2003;301:640-643.
67 Kawai T, Akira S. Toll-like receptor and RIG-I-like receptor signaling. Ann N Y Acad Sci 2008;1143:1-20.
68 Cavanaugh PF Jr, Ho YK, Bardos TJ. The activation of murine macrophages and natural killer cells by the partially thiolated double stranded RNA poly(I)-mercapto poly(C). Res Commun Mol Pathol Pharmacol 1996;91:131-147.
69 Schwabe RF, Seki E, Brenner DA. Toll-like receptor signaling in the liver. Gastroenterology 2006;130:1886-1900.
70 Dejager L, Libert C. Tumor necrosis factor alpha mediates the lethal hepatotoxic effects of poly(I:C) in D-galactosaminesensitized mice. Cytokine 2008;42:55-61.
71 Bao Y, Zheng J, Han C, Jin J, Han H, Liu Y, et al. Tyrosine kinase Btk is required for NK cell activation. J Biol Chem 2012;287:23769-23778.
72 He J, Lang G, Ding S, Li L. Pathological role of interleukin-17 in poly I:C-induced hepatitis. PLoS One 2013;8:e73909.
73 Arshad MI, Patrat-Delon S, Piquet-Pellorce C, L’helgoualc’h A, Rauch M, Genet V, et al. Pathogenic mouse hepatitis virus or poly(I:C) induce IL-33 in hepatocytes in murine models of hepatitis. PLoS One 2013;8:e74278.
74 Hou X, Song J, Su J, Huang D, Gao W, Yan J, et al. CD4(+)Foxp3(+) Tregs protect against innate immune cell-mediated fulminant hepatitis in mice. Mol Immunol 2015;63:420-427.
75 Jiang W, Sun R, Wei H, Tian Z. Toll-like receptor 3 ligand attenuates LPS-induced liver injury by down-regulation of tolllike receptor 4 expression on macrophages. Proc Natl Acad Sci U S A 2005;102:17077-17082.
76 Olleros ML, Vesin D, Fotio AL, Santiago-Raber ML, Tauzin S, Szymkowski DE, et al. Soluble TNF, but not membrane TNF, is critical in LPS-induced hepatitis. J Hepatol 2010;53:1059-1068.
77 Gantner F, Leist M, Lohse AW, Germann PG, Tiegs G. Concanavalin A-induced T-cell-mediated hepatic injury in mice: the role of tumor necrosis factor. Hepatology 1995;21:190-198.
78 Sass G, Shembade ND, Haimerl F, Lamoureux N, Hashemolhosseini S, Tannapfel A, et al. TNF pretreatment interferes with mitochondrial apoptosis in the mouse liver by A20-mediated down-regulation of Bax. J Immunol 2007;179:7042-7049.
79 Kuhla A, Eipel C, Abshagen K, Siebert N, Menger MD, Vollmar B. Role of the perforin/granzyme cell death pathway in D-Gal/ LPS-induced inflammatory liver injury. Am J Physiol Gastrointest Liver Physiol 2009;296:G1069-1076.
80 Dong Z, Wei H, Sun R, Tian Z. The roles of innate immune cells in liver injury and regeneration. Cell Mol Immunol 2007;4:241-252.
81 Corazza N, Badmann A, Lauer C. Immune cell-mediated liver injury. Semin Immunopathol 2009;31:267-277.
82 Wroblewski R, Armaka M, Kondylis V, Pasparakis M, Walczak H, Mittrücker HW, et al. Opposing role of tumor necrosis factor receptor 1 signaling in T cell-mediated hepatitis and bacterial infection in mice. Hepatology 2016;64:508-521.
83 Yang P, Zhou W, Li C, Zhang M, Jiang Y, Jiang R, et al. Kupffercell-expressed transmembrane TNF-α is a major contributor to lipopolysaccharide and D-galactosamine-induced liver injury. Cell Tissue Res 2016;363:371-383.
84 Furuya S, Kono H, Hara M, Hirayama K, Sun C, Fujii H. Interleukin 17A plays a role in lipopolysaccharide/D-galactosamine-induced fulminant hepatic injury in mice. J Surg Res 2015;199:487-493.
85 Chen Z, Liu H, Lei S, Zhao B, Xia Z. LY294002 prevents lipopolysaccharide induced hepatitis in a murine model by suppressing IκB phosphorylation. Mol Med Rep 2016;13:811-816.
86 Homberger FR. Enterotropic mouse hepatitis virus. Lab Anim 1997;31:97-115.
87 Haring J, Perlman S. Mouse hepatitis virus. Curr Opin Microbiol 2001;4:462-466.
88 Jacques A, Bleau C, Martin JP, Lamontagne L. Intrahepatic endothelial and Kupffer cells involved in immunosuppressive cytokines and natural killer (NK)/NK T cell disorders in viral acute hepatitis. Clin Exp Immunol 2008;152:298-310.
89 Yang W, Ding X, Deng J, Lu Y, Matsuda Z, Thiel A, et al. Interferon-gamma negatively regulates Th17-mediated immunopathology during mouse hepatitis virus infection. J Mol Med (Berl) 2011;89:399-409.
90 Chen Y, Wu S, Guo G, Fei L, Guo S, Yang C, et al. Programmed death (PD)-1-deficient mice are extremely sensitive to murine hepatitis virus strain-3 (MHV-3) infection. PLoS Pathog 2011;7:e1001347.
91 Aparicio JL, Peña C, Retegui LA. Autoimmune hepatitis-like disease in C57BL/6 mice infected with mouse hepatitis virus A59. Int Immunopharmacol 2011;11:1591-1598.
92 Lamontagne L, Descoteaux JP, Jolicoeur P. Mouse hepatitis virus 3 replication in T and B lymphocytes correlate with viral pathogenicity. J Immunol 1989;142:4458-4465.
93 Bleau C, Filliol A, Samson M, Lamontagne L. Brain invasion by mouse hepatitis virus depends on impairment of tight junctions and beta interferon production in brain microvascular endothelial cells. J Virol 2015;89:9896-9908.
94 Marro BS, Grist JJ, Lane TE. Inducible expression of CXCL1 within the central nervous system amplifies viral-induced demyelination. J Immunol 2016;196:1855-1864.
95 Teoh NC. Hepatic ischemia reperfusion injury: contempo-rary perspectives on pathogenic mechanisms and basis for hepatoprotection-the good, bad and deadly. J Gastroenterol Hepatol 2011;26:180-187.
96 Zorde-Khvalevsky E, Abramovitch R, Barash H, Spivak-Pohis I, Rivkin L, Rachmilewitz J, et al. Toll-like receptor 3 signaling attenuates liver regeneration. Hepatology 2009;50:198-206.
97 Enkhbold C, Morine Y, Utsunomiya T, Imura S, Ikemoto T, Arakawa Y, et al. Dysfunction of liver regeneration in aged liver after partial hepatectomy. J Gastroenterol Hepatol 2015;30:1217-1224.
98 Luedde T, Kaplowitz N, Schwabe RF. Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterology 2014;147:765-783.e4.
99 Baeck C, Tacke F. Balance of inflammatory pathways and interplay of immune cells in the liver during homeostasis and injury. EXCLI J 2014;13:67-81.
100 Fischer R, Baumert T, Blum HE. Hepatitis C virus infection and apoptosis. World J Gastroenterol 2007;13:4865-4872.
101 Bantel H, Lügering A, Poremba C, Lügering N, Held J, Domschke W, et al. Caspase activation correlates with the degree of inflammatory liver injury in chronic hepatitis C virus infection. Hepatology 2001;34:758-767.
102 Gao B, Radaeva S, Park O. Liver natural killer and natural killer T cells: immunobiology and emerging roles in liver diseases. J Leukoc Biol 2009;86:513-528.
103 Dunn C, Brunetto M, Reynolds G, Christophides T, Kennedy PT, Lampertico P, et al. Cytokines induced during chronic hepatitis B virus infection promote a pathway for NK cellmediated liver damage. J Exp Med 2007;204:667-680.
104 Rehermann B. Pathogenesis of chronic viral hepatitis: differential roles of T cells and NK cells. Nat Med 2013;19:859-868.
105 Yuan L, Kaplowitz N. Mechanisms of drug-induced liver injury. Clin Liver Dis 2013;17:507-518, vii.
106 Natori S, Rust C, Stadheim LM, Srinivasan A, Burgart LJ, Gores GJ. Hepatocyte apoptosis is a pathologic feature of human alcoholic hepatitis. J Hepatol 2001;34:248-253.
107 Nagy LE. The Role of innate immunity in alcoholic liver disease. Alcohol Res 2015;37:237-250.
108 Gautheron J, Vucur M, Reisinger F, Cardenas DV, Roderburg C, Koppe C, et al. A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol Med 2014;6:1062-1074.
109 Casey CA, Nanji A, Cederbaum AI, Adachi M, Takahashi T. Alcoholic liver disease and apoptosis. Alcohol Clin Exp Res 2001;25:49S-53S.
110 Ziol M, Tepper M, Lohez M, Arcangeli G, Ganne N, Christidis C, et al. Clinical and biological relevance of hepatocyte apoptosis in alcoholic hepatitis. J Hepatol 2001;34:254-260.
June 15, 2016
Accepted after revision November 11, 2016
Author Affiliations: Institute of Microbiology (Khan HA and Arshad MI), and Institute of Pharmacy, Physiology and Pharmacology (Khan JA), University of Agriculture, Faisalabad, Pakistan; Department of Pathology, The University of Faisalabad (TUF), Faisalabad, Pakistan (Ahmad MZ)
Muhammad Imran Arshad, PhD, Assistant Professor, Institute of Microbiology, University of Agriculture, Faisalabad 38040, Pakistan (Tel: +92-41-9200161ext3113; Email: drimranarshad@ yahoo.com)
© 2017, Hepatobiliary Pancreat Dis Int. All rights reserved.
10.1016/S1499-3872(17)60014-6
Published online April 24, 2017.
杂志排行

Hepatobiliary & Pancreatic Diseases International的其它文章
- Hepatobiliary & Pancreatic Diseases International
- Pancreatic head excavation for tissue diagnosis may reduce unnecessary pancreaticoduodenectomies in the setting of chronic pancreatitis
- Combined cavo-atrial thrombectomy and hepatectomy in hepatocellular carcinoma
- Traditional surgical planning of liver surgery is modified by 3D interactive quantitative surgical planning approach: a single-center experience with 305 patients
- A clinical analysis of acute pancreatitis in pregnancy
- Circulating autoantibodies to endogenous erythropoietin are associated with chronic hepatitis C virus infection-related anemia