乳酸克鲁维酵母超量表达葡萄糖氧化酶的研究
2017-06-01李国丽翟立翔李师翁陈熙明
李 娟,李国丽,翟立翔,李师翁,*,陈熙明
(1.兰州交通大学化学与生物工程学院,甘肃省极端环境微生物资源与工程重点实验室,甘肃兰州 730070;.中国科学院西北生态环境研究院沙漠与沙漠化重点实验室,甘肃兰州 730000)
乳酸克鲁维酵母超量表达葡萄糖氧化酶的研究
李 娟1,李国丽1,翟立翔1,李师翁1,*,陈熙明2
(1.兰州交通大学化学与生物工程学院,甘肃省极端环境微生物资源与工程重点实验室,甘肃兰州 730070;.中国科学院西北生态环境研究院沙漠与沙漠化重点实验室,甘肃兰州 730000)
本研究应用具有超量分泌表达能力的乳酸克鲁维酵母(KlSEL1基因突变型)、游离型载体pKDU7以及整合型表达载体pKLAC1,对具有5个氨基酸点突变的葡萄糖氧化酶(该突变酶的比活是野生型葡萄糖氧化酶的3.24倍)进行分泌表达。最终获得了一株可以超量分泌表达葡萄糖氧化酶的菌株,其最大产量为70±7 kU/L。这是现今已报道的分泌表达葡萄糖氧化酶能力最高的乳酸克鲁维重组菌株,为食品安全级的葡萄糖氧化酶的生产和应用提供了新途径。
葡萄糖氧化酶,乳酸克鲁维酵母,表达
葡萄糖氧化酶能够在有氧气的条件下专一性地催化D-葡萄糖生成过氧化氢和葡萄糖酸。这类酶最早在黑曲霉中被发现并广泛应用。例如被用作面粉添加剂和食品保鲜剂[1],用来生产葡萄糖酸[2]以及作为肠道微生物菌群调节剂[3]。工业化生产中常用黑曲霉和青霉发酵来生产葡萄糖氧化酶,然而在发酵过程中常会伴随产生过氧化氢酶、淀粉酶等其他杂蛋白[4-5],给后期的纯化工作带来很大的困难,利用基因工程菌株重组表达有利于解决这一难题。前人已在大肠杆菌表达系统中成功表达了葡萄糖氧化酶[6],但其表达产物无翻译后的加工与修饰过程,不能对蛋白质进行翻译。利用毕赤酵母表达系统可对蛋白进行翻译,然而毕赤酵母并不是食品安全级酵母,不能应用于食品行业。
乳酸克鲁维酵母为美国FDA认证的食品安全级酵母[7],其表达系统具有以下优点:乳酸克鲁维酵母表达的产物无需复杂的纯化过程,可直接应用于食品及饲料工业中;发酵过程无产气现象,不会发生爆罐;在发酵过程中所需的诱导剂是乳糖或半乳糖,所以更安全;可用来高密度发酵。然而该系统的缺点是蛋白表达量相对毕赤酵母发酵系统较低。前人利用毕赤酵母作为宿主对葡萄糖氧化酶进行异源表达获得40 kU/L葡萄糖氧化酶的产量[8],而利用乳酸克鲁维酵母为宿主对葡萄糖氧化酶进行异源表达的葡萄糖氧化酶产量仅为2 kU/L[9]。

表1 不同表达载体的扩增条件
在乳酸克鲁维酵母中有许多限制因子,如抑制分泌表达的基因KlSEL1,该基因的突变可以有效提升蛋白分泌表达能力5倍左右[10],所以该突变更有利于葡萄糖氧化酶的表达。另外在酵母中提高目的基因的拷贝数也可以有效提高蛋白的表达水平。本研究应用具有超量分泌表达能力的乳酸克鲁维酵母(KlSEL1基因突变型)游离型载体pKDU7[11]和整合型表达载体pKLAC1,对具有5个氨基酸点突变的葡萄糖氧化酶(N2Y、K13E、T30V、I94V和K152R)进行了分泌表达。
1 材料与方法
1.1 材料与仪器
1.1.1 菌株和质粒 大肠杆菌(Escherichiacoli)DH52(菌株由本实验室保存),乳酸克鲁维酵母(KlSEL1基因突变型)MD2/1菌株和游离型载体pKLDU7由威尼斯Vilniu大学生物技术研究所Kestuis Sasnauskas博士惠赠。pKLAC1载体购自NEB公司。
Pfu DNA Polymerase 大连宝生物(TaKaRa)生物工程有限公司;DNA片段回收试剂盒 百泰克生物工程公司;PCR引物 由上海生工生物工程有限公司合成;基因测序 由北京六合华大基因有限公司完成;限制性内切酶、T4DNA 连接酶 购自NEB公司。其他化学试剂均为国产分析纯。
1.1.2 仪器 DNA电泳仪JY3000 北京君逸电泳仪公司;SA-1000凝胶成像系统 美国Alpha Innotech上海吉泰依科赛生物科技有限公司;分析天平FA1004N 上海精科仪器有限公司;小型垂直蛋白电泳仪 美国Bio-Rad公司;气浴恒温振荡器THZ-82B 江苏正基仪器有限公司。
1.2 实验方法
1.2.1 培养基 LB培养基(1L):10 g胰蛋白胨,5 g酵母提取物,10 g NaCl。
YPD培养基(1L):20 g胰蛋白胨,10 g酵母提取物,20 g葡萄糖。
YPGal培养基(1L):20 g胰蛋白胨,10 g酵母提取物,20 g半乳糖。
YCB培养基(1L):5 mmol/L酰胺,1%葡萄糖,0.1% KH2PO4,0.05% MgSO4,0.01% NaCl,0.01% KCl,1 mmol/L精氨酸,1 mmol/L赖氨酸,微量元素及维生素。
YDFM培养基(1L):10 g硫酸铵,30 g蔗糖,1 mmol/L精氨酸,1 mmol/L赖氨酸,11.83 g KH2PO4,2.29 g K2HPO4,1 g MgSO4,0.33 g CaCl2,1 g NaCl,1 g KCl,微量元素及维生素[13]。
1.2.2 葡萄糖氧化酶基因的合成与整合型质粒pKLACGOX的构建 野生型葡萄糖氧化酶首先是在黑曲霉(Aspergillusniger)中发现并报道。该酶基因全长1749 bp,编码583个氨基酸。根据Prodanovic R等的报道[12],葡萄糖氧化酶中5个氨基酸的点突变(N2Y、K13E、T30V、I94V和K152R)可提高该酶的活性3.24倍。因此,我们根据乳酸克鲁维酵母的密码子使用偏好,我们采用全基因合成的方式,对点突变后的葡萄糖氧化酶所对应的DNA序列进行了合成。
为构建pKLACGOX表达载体,根据合成的葡萄糖氧化酶基因序列,设计引物pKLAC-F(5′-CACCTCGAGAAAAGATCCTATGGTATTGAAGC-3′)和pKLAC-R(5′-GTCGGTACCTTATTGCATACTT GCGTAATCTTCC-3′),在上下游引物中分别引入XhoI和KpnI酶切位点。以合成的葡萄糖氧化酶基因质粒DNA为模板,使用pKLAC-F和pKLAC-R进行PCR扩增。PCR扩增条件见表1。用胶回收试剂盒回收扩增片段。回收后的扩增片段与PMD18T载体进行连接,连接产物转化DH5a大肠杆菌后挑取阳性克隆进行测序。
测序验证后的阳性克隆,在LB培养基中进行扩繁,并提取质粒使用XhoI和KpnI进行双酶切,酶切后使用1%的琼脂糖凝胶电泳对目的片段进行检测和回收。为使回收后的葡萄糖氧化酶片段插入pKLAC1载体,对pKLAC1载体同样使用XhoI和KpnI进行了双酶切,琼脂糖凝胶电泳后对载体片段进行了回收。回收后的葡萄糖氧化酶片段和pKLAC1载体使用T4连接酶16 ℃过夜连接后,转化DH5a后使用菌落PCR方法鉴定阳性克隆。
1.2.3 游离型质粒pKLDUGOX的构建 用SacII限制性内切酶对重组质粒pKLACGOX进行酶切,将酶切后的较大片段(6430 bp)使用凝胶回收试剂盒进行回收待用。通过化学转化法将酶切后的大片段DNA转化到乳酸克鲁维酵母MD2/1中。在YCM培养基中筛选获得阳性转化子。通过液体YPD对阳性转化子进行培养,并利用酵母DNA提取试剂盒提取DNA。以酵母基因组为模板,利用引物P1:GTCAGTGGGCCGAAATATG和P2:ATACAACATCG AAGAAGAGTCTTTCTTTAGG对其中整合的PLAC4-PBI-GOX进行扩增。PCR扩增条件见表1。用胶回收试剂盒回收扩增片段。将游离型质粒pKDU7用SmaI进行酶切,37 ℃条件下1 h。将从酵母中扩增出的PLAC4-PBI-GOX片段和酶切好的质粒pKDU7用T4DNA连接酶16 ℃连接过夜。将连接产物转化到大肠杆菌感受态中,在含氨苄青霉素的LB平板上筛选阳性克隆。挑取阳性菌落扩大培养,提取质粒后分别进行菌落PCR。PCR鉴定正确的阳性质粒送往华大测序,用来确认序列和读码框,最后验证无误的重组质粒命名为pKLDUGOX。
1.2.4 整合型质粒pKLACGOX的转化 用SacII限制性内切酶将pKLACGOX质粒进行线性化,并浓缩回收。挑取乳酸克鲁维酵母(KlSEL1基因突变型)MD2/1的单菌落,将其接种到10 mL的YPD培养基中,30 ℃摇菌过夜。使用化学转化法将线性化的质粒转到乳酸克鲁维酵母(KlSEL1基因突变型)MD2/1中,在含有尿嘧啶的YCB培养基平板上30 ℃培养4 d后进行鉴定。具体转化方法见参考文献[14]。由于受体菌乳酸克鲁维酵母MD2/1既是尿嘧啶缺陷型,又是KlSEL1基因突变型,pKLAC1载体携带乙酰胺降解基因,因此只有线性化pKLACGOX片段整合到酵母基因组中才能在含有尿嘧啶的YCB平板上生长。挑取长出来的单菌落进行摇瓶培养,培养好的菌一部分用来保存菌种,命名为MD2/1-pKLACGOX,另一部分用来提DNA。用同上转化方法将质粒pKLAC1转到乳酸克鲁维酵母(KlSEL1基因突变型)菌株MD2/1中,命名为MD2/1-pKLAC1菌株。
1.2.5 游离型质粒pKLDUGOX的转化 挑取MD2/1-pKLACGOX的单菌落,将其接种到10 mL的YPD培养基中,30 ℃摇菌过夜。之后转化方法与将pKLACGOX质粒转化到菌株MD2/1中相同。取200 μL菌液涂布于YCB培养基平板上,30 ℃培养4 d后进行鉴定。由于受体菌MD2/1-pKLACGOX携带乙酰胺降解基因,pKDU7载体携带尿嘧啶合成代谢基因,因此只有转化成功的阳性克隆才能在不含尿嘧啶的YCB平板上生长。在YCB平板上生长出来的单菌落既是阳性转化子。挑取长出来的单菌落在YPD培养基中进行摇瓶培养,取一部分培养好的菌进行菌种保存,命名为MD2/1-15BGOX,另一部分用来提DNA。用同上转化方法将质粒pKDU7转化到MD2/1-pKLAC1菌株中,即为对照菌株,命名为MD2/1-15B。
1.2.6 乳酸克鲁维酵母工程菌的诱导表达 将工程菌株MD2/1-15BGOX接种于20 mL YPGal的三角瓶中,30 ℃,220 r/min在摇床中进行培养,分别在0、24、48、72、96、120、144、168 h取上清1 mL离心,收集上清并保存。另外同样将工程菌株MD2/1-15BGOX接种于20 mL YDFM为培养基的三角瓶中,30 ℃,220 r/min在摇床中进行培养,每24 h补一次蔗糖,共补蔗糖40 g/L,之后再用终浓度为20 g/L的半乳糖进行诱导,分别在0、24、48、72、96、120、144和168 h取上清1 mL离心,收集上清并保存。
1.2.7 不同诱导时间表达产物的分析和鉴定 将上清液进行酶活测定和SDS-PAGE的分析,检测重组蛋白的表达含量。根据诱导的不同时间测定葡萄糖氧化酶的活性,测定方法如下:在100 μL体系中加入10 μL 1 mol/L NaH2PO4,pH6.0,10 μL 100 mmol/L葡萄糖,0.75 mg/mL邻联茴香胺,0.1 U辣根过氧化物酶以及10 μL葡萄糖氧化酶液体,并用去离子水补至100 μL。于30 ℃反应30 min后加入134 μL 12 mol/L的硫酸反应,540 nm测定吸光值。不同时间酶活测定分别测3个平行。葡萄糖氧化酶酶活定义:pH6.0、30 ℃的条件下,每分钟能把 1.0 μmol的β-D-葡萄糖氧化成D-葡萄糖酸和H2O2的酶量为一个单位。SDS-PAGE的分析方法详见[15]。
1.2.8 不同温度在不同时间下对葡萄糖氧化酶酶活影响的测定 首先将葡萄糖氧化酶液体各取100 μL分别放在37、42、50、60 ℃的水浴锅中,在1、2、3、4 h后分别测其酶活,每个测3个平行。测定方法:同1.2.6。
1.2.9 pH对葡萄糖氧化酶酶活影响的测定 测定方法如下:在100 μL体系中加入10 μL1 mol/L的不同pH的缓冲液,10 μL 100 mmol/L葡萄糖,0.75 mg/mL邻联茴香胺,0.1 U辣根过氧化物酶以及10 μL葡萄糖氧化酶液体,并用去离子水补至100 μL。于30 ℃反应30 min后加入134 μL 12 mol/L的硫酸反应,540 nm测定吸光值,各测3个平行。不同pH的缓冲液为:3.0、4.0、5.0(50 mmol/L乙酸盐);6.0、7.0、8.0(50 mmol/L 羟乙基哌嗪乙磺酸);9.0、10.0、11.0、12.0(50 mmol/L Tris)[7]。
1.2.10 不同NaCl浓度对葡萄糖氧化酶酶活影响的测定 测定方法如下:在100 μL体系中加入10 μL 1 mol/L NaH2PO4,pH6.0,10 μL 100 mmol/L葡萄糖,0.75 mg/mL邻联茴香胺,0.1 U辣根过氧化物酶以及10 μL葡萄糖氧化酶液体,并用不同盐浓度溶液补至100 μL。于30 ℃反应30 min后加入134 μL 12 mol/L的硫酸反应,540 nm测定吸光值,各测3个平行。不同NaCl浓度分别为:50、100、200、300、400、500、600、750、1000、1200、1500、2000 mmol/L。
1.3 数据统计分析
实验数据每个3个平行,利用Origin 7.5软件处理数据作图,并用SAS 8.01进行统计分析,进行t检验,以p<0.05为显著水平。
2 结果与分析
2.1 葡萄糖氧化酶基因表达片段的PCR扩增
通过Pfu DNA Polymerase获得了大小约为1749 bp的扩增片段(图1),这与黑曲霉葡萄糖氧化酶基因序列大小一致。

图1 PCR扩增-葡萄糖氧化酶基因Fig.1 glucose oxidase gene after PCR amplification注:1-DNA Marker;2-目的基因PCR产物。
2.2 重组整合型质粒pKLACGOX的鉴定
如图2所示,葡萄糖氧化酶基因经过PCR扩增后,与整合型表达载体pKLAC1连接,构建成重组质粒pKLACGOX,目标基因之后连接有GOX基因序列。将连接好的表达载体进行PCR扩增鉴定,继而进行酶切鉴定,结果见图3。将该阳性质粒送交北京六合华大基因测序有限公司测序,结果显示基因序列完整,无碱基突变,且重组表达载体的读码框正确,表达载体pKLACGOX构建成功。
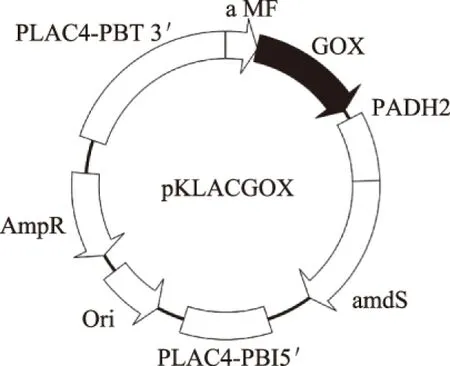
图2 重组质粒pKLACGOX的结构Fig.2 Genetic map of the recombinant plasmid pKLACGOX
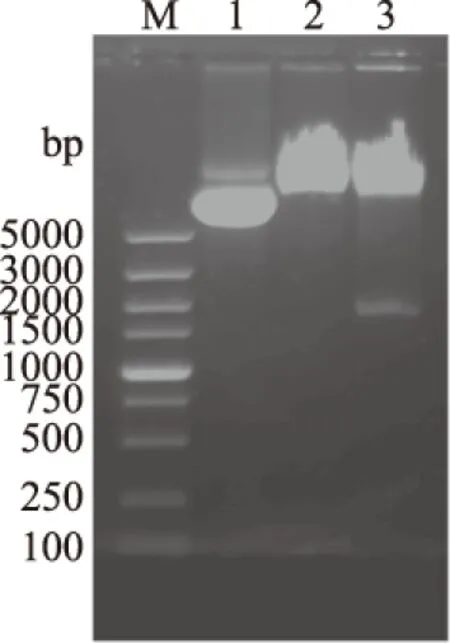
图3 质粒pKLACGOX的酶切鉴定Fig.3 Identification of the plasmid pKLACGOX by endonuclease digestion注:M-DNA Marker;1-质粒pKLACGOX; 2-质粒pKLACGOX经Xho I单酶切后的片段; 3-质粒pKLACGOX经Xho I/Kpn I酶切后的片段。
2.3 重组游离型质粒pKLDUGOX的鉴定
如图4所示,通过将整合有pKLACGOX质粒的阳性酵母菌株的DNA上扩增获得的PLAC4-PBI-GOX通过平末端连接整合入pKDU7中,构建成重组质粒pKLDUGOX。将连接好的表达载体进行PCR扩增鉴定,结果见图5。将该阳性质粒送交北京六合华大基因测序有限公司测序,结果显示基因序列完整,无碱基突变,且重组表达载体的读码框正确,表达载体pKLDUGOX构建成功。
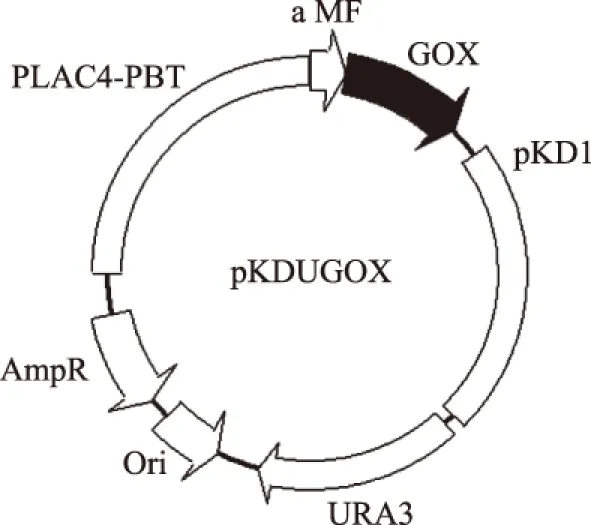
图4 重组质粒pKLDUGOX的结构Fig.4 Genetic map of the recombinant plasmid pKLDUGOX

图5 质粒pKLDUGOX的PCR扩增鉴定Fig.5 Determination of the plasmid pKLDUGOX using PCR amplictiaon注:1-DNA Marker;2-用阳性转化子为模板对插入片段扩增; 3-用阴性对照-质粒pKLDU7为模板对插入片段扩增。
2.4 化学法转化乳酸克鲁维酵母阳性的筛选
为了进一步鉴定阳性菌株能否产葡萄糖氧化酶,我们在含有葡萄糖、乳糖、邻联茴香胺及辣根过氧化物酶的固体培养基中对两种菌株进行培养,仅在含有葡萄糖氧化酶表达菌株处出现了棕色(图6),而该颜色为表达的葡萄糖氧化酶催化葡萄糖与氧气形成的过氧化氢与邻联茴香胺受到辣根过氧化物酶偶联催化形成的显色反应。

图6 重组葡萄糖氧化酶菌株的显色反应鉴定Fig.6 Identification of the glucose oxidase recombinant strain using the chromogenic reaction注:左图-对照菌株,右图-阳性菌株。
2.5 葡萄糖氧化酶表达活性检测及表达量分析
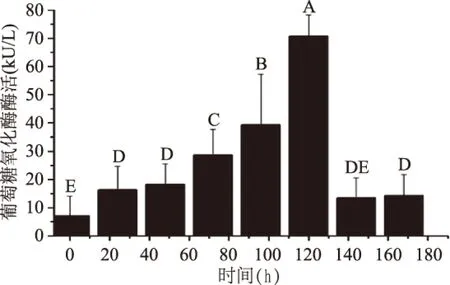
图7 重组菌株摇瓶发酵培养中葡萄糖氧化酶的酶活的变化Fig.7 The glucose oxidase activity induced by galactose during flask-shaking fermentation of the recombinant strain
为了进一步了解工程菌株MD2/1-15BGOX对葡萄糖氧化酶重组表达的能力,将菌株MD2/1-15BGOX在不同诱导时期进行取样。图7表明随着诱导时间的延长发酵液中的葡萄糖氧化酶活性逐渐上升,在诱导72 h后葡萄糖氧化酶活性出现快速升高。在120 h后发酵上清液中葡萄糖氧化酶活性达到最高为(70±7) kU/L。由t检验可知,p<0.05。图7中不同字母表示处理间的差异具有显著性,显著性大小依次是A>B>C>D>E,标有字母DE表示差异不显著。图8中,我们利用SDS-PAGE分析上清中蛋白表达量的变化,在葡萄糖氧化酶理论分子量66 kDa处并未出现高表达的特异条带,然而在分子量>110 kDa(*)条带处随着诱导时间的延长条带颜色逐渐加深,这与图7呈现的结果一致。也与Marija Blazic[14]等人报道的用酿酒酵母表达葡萄糖氧化酶的分子量的结果相符合。
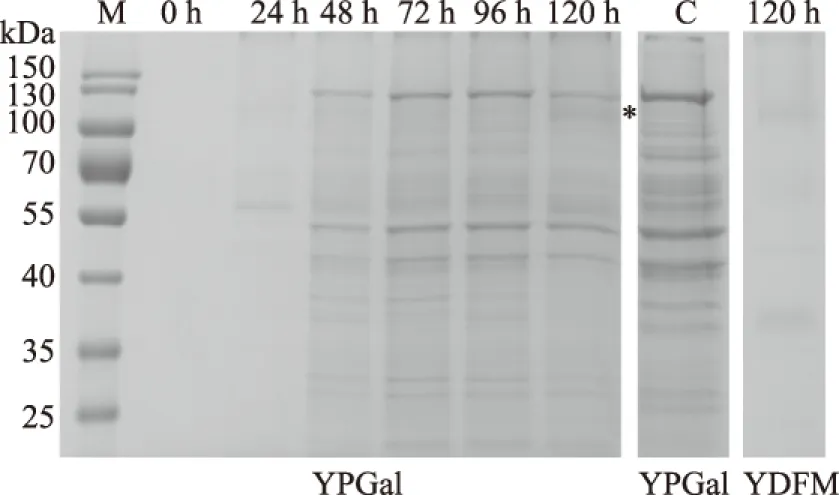
图8 摇瓶发酵液SDS-PAGE分析Fig.8 SDS-PAGE analysis of the supernatant from the flask-shaking fermentation of the recombinant strain
为了去除由于培养基中胰蛋白胨、酵母提取物对乳酸克鲁维酵母分泌表达蛋白的影响,我们使用了YDFM培养基对MD2/1-15BGOX进行了培养及诱导表达。YDFM是一种仅由蔗糖与硫酸铵为唯一碳氮源的酵母发酵培养基。利用YDFM对MD2/1-15BGOX培养至对数生长中后期,加入终浓度为20 g/L的半乳糖对葡萄糖氧化酶进行诱导表达。尽管随着诱导时间的延长发酵液中的葡萄糖氧化酶活性也在逐渐上升,但是相比在YPGal中葡萄糖氧化酶活性却低得多,诱导120 h后上清的酶活仅有16 kU/L。通过SDS-PAGE分析发现MD2/1-15BGOX在YDFM中表达蛋白的种类相对较少,仅在分子量>110 kDa处有一条特异条带其余蛋白条带均小于葡萄糖氧化酶预计蛋白分子量(图8)。因此说明乳酸克鲁维酵母表达的葡萄糖是糖基化程度较高的蛋白,且该蛋白具有良好的葡萄糖氧化酶活性。
2.6 pH、温度和盐浓度对葡萄糖氧化酶酶活的影响
我们研究了不同pH、不同温度、不同NaCl浓度胁迫对葡萄糖氧化酶酶活的影响。从图9中可以看出最大酶活在pH为4时,达(79.45±11) kU/L。葡萄糖氧化酶工作的正常pH范围为4~7,这个结果与之前的报道是相符的[9,17]。在碱性pH下酶活很低,图10表明,在37 ℃培养4 h期间,酶活都比较稳定。然而随着温度的逐渐上升,葡萄糖氧化酶加速失活,Gouda MD[18]等人研究表明A. niger葡萄糖氧化酶在大于50 ℃后活性丧失。从图11表明,随着盐浓度的升高,酶活逐渐降低,这说明高盐对葡萄糖氧化酶活性具有强烈的抑制作用。
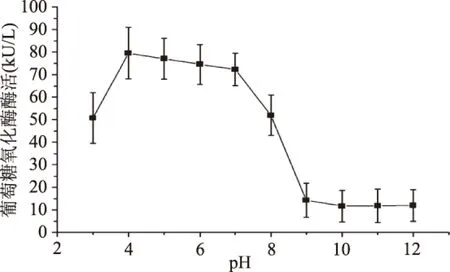
图9 不同pH对葡萄糖氧化酶酶活的影响Fig.9 Effect of pH on the glucose oxidase activity
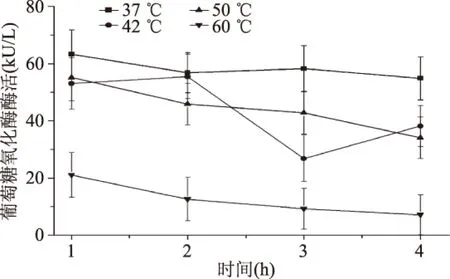
图10 不同温度对葡萄糖氧化酶酶活的影响Fig.10 Effect of temperature on the glucose oxidase activity

图11 不同NaCl浓度对葡萄糖氧化酶酶活的影响Fig.11 Effect of NaCl concentrations on the glucose oxidase activity
3 结论与讨论
葡萄糖氧化酶是一种重要的工业用酶,尽管前人曾利用乳酸克鲁维酵母对葡萄糖氧化酶进行了重组表达,但最大表达量只有2 kU/L,且在这些重组表达的葡萄糖氧化酶中约有21%结合在酵母细胞壁上并未分泌至上清中[9]。相比前人的酵母表达系统,本研究构建的乳酸克鲁维酵母表达系统分泌葡萄糖氧化酶至发酵上清液中的效率高达(70±7) kU/L,是前人报道的35倍。对比分析表明本研究的表达系统具有高的表达效率的原因是:我们既利用了游离表达载体又利用了整合表达载体,在这两种载体的共同存在下可以保证酵母表达过程中的高基因拷贝数及高稳定性;我们利用了强的诱导表达启动子Lac4,该启动子是现今发现在乳酸克鲁维酵母中启动效率最高的诱导型启动子[19]。SDS-PAGE的结果表明在66 kDa处尚未发现特异性的蛋白条带,然而仅在>110 kDa处发现了特异蛋白条带随着诱导时间的延长逐渐增加。该结果与前人对乳酸克鲁维酵母表达葡萄糖氧化酶的研究结果相符,所以我们推测这条>110 kDa的特异蛋白条带为糖基化后的葡萄糖氧化酶[7]。前人对黑曲霉中的葡萄糖氧化酶进行直接提纯研究,发现这些葡萄糖氧化酶具有非常高的比活且具有高分子量的糖基化修饰[8]。这也表明高分子量糖基化修饰葡萄糖氧化酶是影响高效率表达葡萄糖氧化酶的关键。此外,高pH、高温、高盐易造成酶的构象的改变,从而使酶的催化功能减弱甚至丧失。
[1]Kirstein D,Kuhn W. Glucose-oxidase-Properties and application in the food-industry[J]. Lebensmittel Industrie,1981,28:205-208.
[2]Dzyadevych S,Arkhypova V,Soldatkin A,et al. Amperometric enzyme biosensors:past,present and future[J]. Irbm,2008,29:171-180.
[3]Vartiainen J,Rättö M,Paulussen S. Antimicrobial activity of glucose oxidase-immobilized plasma-activated polypropylene films[J]. Packaging Technology and Science,2005,18:243-251.
[4]Ko JH,HahmMS,Kang HA,et al. Secretory expression and purification of Aspergillus niger glucose oxidase inSaccharomycescerevisiaemutant deficient in PMR1gene[J]. Protein Expression and Purification,2002,25:488-493.
[5]Valdivieso-Ugarte M,Ronehel C,Banuelos O,et al. Expression of an Aspergillus niger glucose oxidase inSaccharomycescerevisiaeand its use to optimize fructo-oligosaccharides synthesis[J]. Biotechnol Prog,2006,22:1096-1101.
[6]Szynol A,Soet D,Tuyl E. Bactericidal effects of a fusion protein of llama heavy-chain antibodies coupled to glucose oxidase on oral bacteria[J]. Antimicrob Agent Chemother,2004,48:3390-3395.
[7]Van-Ooyen AJ,Dekker P,Huang M,et al. Heterologous protein production in the yeastKluyveromyceslactis[J]. FEMS Yeast Research,2006,6:381-392.
[8]Guo Y,Lu F,Zhao H,et al. Cloning and heterologous expression of glucose oxidase gene from Aspergillus niger Z-25 in Pichia pastoris[J]. Applied Biochemistry and Biotechnology,2010,162:498-509.
[9]Rocha SN,Abrahao-Neto J,Cerdan ME,et al. Heterologous expression of glucose oxidase in the yeastKluyveromycesmarxianus[J]. Microbial Cell Factories,2010,9:4.
[10]Bartkeviciute D,Sasnauskas K. Studies of yeastKluyveromyceslactismutations conferring super-secretion of recombinant proteins[J]. Yeast,2003,20:1-11.
[11]Chen XJ. Low-and high-copy-number shuttle vectors for replication in the budding yeastKluyveromyceslactis[J]. Gene,1996,172:131-136.
[12]Prodanovic R,Ostafe R,Scacioc A,et al. Ultrahigh-throughput screening system for directed glucose oxidase evolution in yeast cells[J]. Combinatorial Chemistry & High Throughput Screening,2011,14:55-60.
[13]Ganatra MB,Vainauskas S,Hong JM,et al. A set of aspartyl protease-deficient strains for improved expression of heterologous proteins inKluyveromyceslactis[J]. FEMS Yeast Research,2011,11:168-178.
[14]袁伟,柯涛,杜敏华,等. 牛凝乳酶原基因的合成及其在乳酸克鲁维酵母中的表达[J]. 生物工程学报,2010,26(9):1281-1286.
[15]Chen XM,Jiang Y,Li YT,et al. Regulation of expression of trehalose-6-phosphate synthase during cold shock in Arthrobacter strain A3[J]. Extremophiles:life under extreme conditions,2011,15:499-508.
[16]Blazic M,Kovacevic G,Prodanovic O,et al. Yeast surface display for the expression,purification and characterization of wild-type and B11 mutant glucose oxidases[J]. Protein Expression & Purification,2013,89(2):175-80.
[17]Zia MA,Khalil-ur-Rahman,Saeed MK,et al. Thermal characterization of purified glucose oxidase from a newly isolated Aspergillus niger UAF-1[J]. Journal of Clinical Biochemistry and Nutrition,2007,41:132-138.
[18]Gouda MD,Singh SA,Rao AG,et al. Thermal inactivation of glucose oxidase. Mechanism and stabilization using additives[J]. The Journal of biological chemistry,2003,278:24324-24333.
[19]Colussi PA,Taron CH.KluyveromyceslactisLAC4 promoter variants that lack function in bacteria but retain full function in K. lactis[J]. Applied and Environmental Microbiology,2005,71:7092-7098.
全国中文核心期刊
轻工行业优秀期刊
Over-expression of glucose oxidase gene in the YeastKluyveromyceslactisKlSEL1 strain
LI Juan1,LI Guo-li1,ZHAI Li-xiang1,LI Shi-weng1,*,CHEN Xi-ming2
(1.School of Chemical and Biological Engineering,Lanzhou Jiaotong University;Key Laboratory of Extreme Environmental Microbial Resources and Engineering of Gansu Province,Lanzhou 730070,China;2.Northwest Institute of Eco-Environment and Resources,Chinese Academy of Sciences,Lanzhou 730000,China)
In this study,the glucose oxidase gene with five amino acid sites-directed mutagenesis was synthesized. The activity of this mutant enzyme was 3.24 times than that of the wild type enzyme. The episomal vector pKDU7 and integrative vector pKLAC1 in which the glucose oxidas gene was recombined was constructed. Using these two vectors,the glucose oxidase gene was successfully recombined intoK.lactisKlSEL1 strain and a recombinant strain with efficient secretory over-expression of glucose oxidase was obtained. The result of flask-shaking fermentation showed that the production of glucose oxidase reached 70±7 kU/L in the recombinant strain. This is the highest glucose oxidase-producing recombinantK.lactisstrain reported so far. This technology will provide a promising approach for the production and application of glucose oxidase in food industry.
glucose oxidase;Kluyveromyceslactis;expression
2016-09-28
李娟(1990-),女,硕士研究生,研究方向:主要从事环境微生物学研究,E-mail:18293130625@163.com。
*通讯作者:李师翁(1964-),男,博士,教授,主要从事环境微生物学研究,E-mail:lishweng@mail.lzjtu.cn。
国家自然基金(31400437)资助。
TS202.3
A
1002-0306(2017)05-0168-06
10.13386/j.issn1002-0306.2017.05.023
